
目录
- 1. 原版
- 2. Python版本
- 安装
- 运行
- 原因分析
- 后续分析推荐
1. 原版
安装比较简单。
wget https://reich.hms.harvard.edu/sites/reich.hms.harvard.edu/files/inline-files/XPCLR.tar
tar xvf XPCLR.tar直接运行bin下的XPCLR即可,若不能运行,则编译下:
cd src
make
make install原版分析时准备文件过程较为繁琐,因此更建议使用Python版。
$ /project/biosoft/XPCLR/bin/XPCLR -h
Usage:
XPCLR -xpclr hapmapInput1 hapmapInput2 mapInput outFile -w gWin(Morgan) snpWin gridSize(bp) chrN -p corrLevel
-w1: gWin sets the size of a sliding window(units: 100cM),sWin sets # of SNPs in a window. otherwise, no sliding window
-p1:the input genotpe is already phased. -p0: the input genotype is not phased
corrLevel: the value is on (0,1], set corrLevel equal to 0 if no correction is needed2. Python版本
安装
conda create -n xpclr -c bioconda xpclr
conda activate xpclr报错:
$ xpclr -h
Traceback (most recent call last):
File "/home/miniconda3/envs/xpclr/bin/xp服务器托管网clr", line 5, in
import xpclr
File "/home/miniconda3/envs/xpclr/lib/python2.7/site-packages/xpclr/__init__.py", line 3, in
from xpclr import methods
File "/home/miniconda3/envs/xpclr/lib/python2.7/site-packages/xpclr/methods.py", line 11, in
from functools import lru_cache
ImportError: cannot import name lru_cache重新安装pip install lru_cache仍然报错。
由于python版本引起,直接修改methods.py中from functools import lru_cache的为:
try:
from functools import lru_cache
except ImportError:
from backports.functools_lru_cache import lru_cache运行
xpclr --format vcf --input /project/04.sweep/sample750_miss0.6_impute/meanDP3.miss0.6.maf0.01.impute.rename.vcf
--samplesA /project/04.sweep/sample750_miss0.6_impute/List/w-l-c/Cultivar.list
--samplesB /project/04.sweep/sample750_miss0.6_impute/List/w-l-c/Wild.list
--chr 1 --maxsnps 600 --size 1000 --step 1000 --out test_out报错:
Traceback (most recent call last):
File "/home/miniconda3/envs/xpclr/bin/xpclr", line 195, in
main()
File "/home/miniconda3/envs/xpclr/bin/xpclr", line 88, in main
"No permission to write in the specified directory: {0}".format(outdir)
AssertionError: No permission to write in the specified directory:xpclr第84行fn = args.out修改为:
fn = os.path.abspath(args.out)运行继续报错:
2023-03-17 19:05:48 : INFO : running xpclr v1.1.0
2023-03-17 19:05:48 : INFO : Loading VCF
Traceback (most recent call last):
File "/home/miniconda3/envs/xpclr/bin/xpclr", line 196, in
main()
File "/home/miniconda3/envs/xpclr/bin/xpclr", line 103, in main
gdistkey=args.gdistkey)
File "/home/miniconda3/envs/xpclr/lib/python2.7/site-packages/xpclr/util.py", line 112, inload_vcf_format_data
pos1, geno1 = load_vcf_wrapper(vcf_fn, chrom, samples1)
File "/ho服务器托管网me/miniconda3/envs/xpclr/lib/python2.7/site-packages/xpclr/util.py", line 94, in load_vcf_wrapper
callset = allel.read_vcf(
AttributeError: 'module' object has no attribute 'read_vcf'网上没有我的同类安装相关错误报道,查看了下allel模块,确实没有read_vcf函数:
Python 2.7.18 |Anaconda, Inc.| (default, Nov 25 2022, 06:27:37)
[GCC 11.2.0] on linux2
Type "help", "copyright", "credits" or "license" for more information.
>>> import allel
>>> allel
>>> dir(allel)
['AlleleCountsArray', 'AlleleCountsCArray', 'AlleleCountsCTable', 'AlleleCountsChunkedArray', 'AlleleCountsChunkedTable', 'AlleleCountsDaskArray', 'FeatureCTable', 'FeatureChunkedTable', 'FeatureTable', 'GenotypeArray', 'GenotypeCArray', 'GenotypeChunkedArray', 'GenotypeDaskArray', 'HaplotypeArray', 'HaplotypeCArray', 'HaplotypeChunkedArray', 'HaplotypeDaskArray', 'SortedIndex', 'SortedMultiIndex', 'UniqueIndex', 'VariantCTable', 'VariantChunkedTable', 'VariantTable', '__builtins__', '__doc__', '__file__', '__loader__', '__name__', '__package__', '__path__', '__version__', '_bcolz', '_da', 'chunked', 'compat', 'constants', 'io', 'model', 'plot', 'stats', 'util']但实际上官网上是有这个函数的:
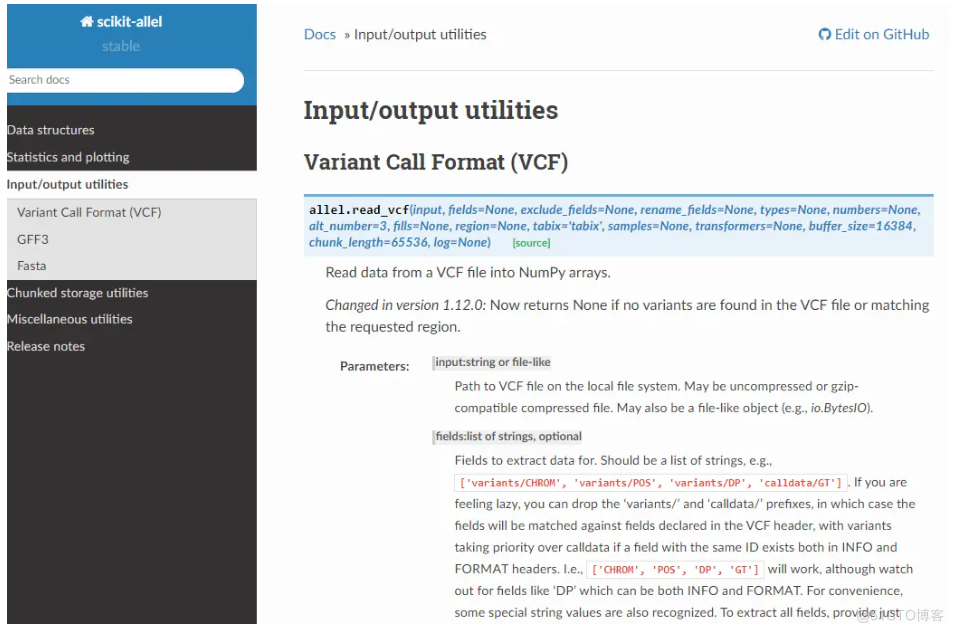
难道是版本问题吗?装了下python3版本,发现确实是有的:
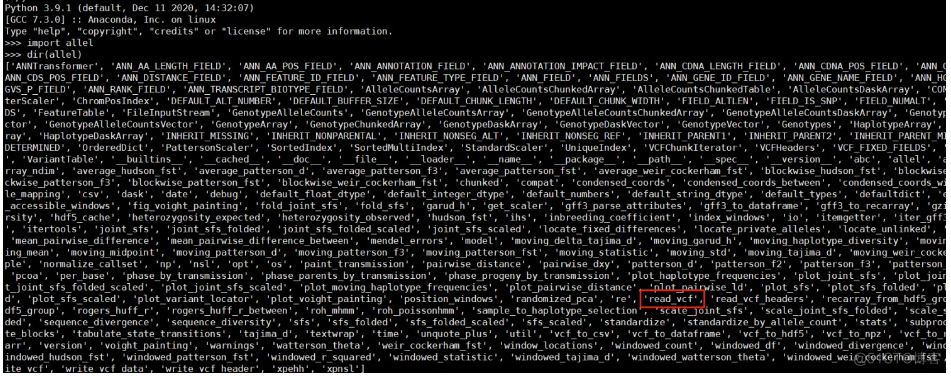
试图重新安装,旧版本删除不了:
$ pip uninstall scikit-allel
DEPRECATION: Python 2.7 will reach the end of its life on January 1st, 2020. Please upgrade your Python as Python 2.7 won't be maintained after that date. A future version of pip will dropsupport for Python 2.7. More details about Python 2 support in pip, can be found at https://pip.pypa.io/en/latest/development/release-process/#python-2-support
ERROR: Cannot remove entries from nonexistent file /home/pengjx/miniconda3/envs/xpclr/lib/python2.7/site-packages/easy-install.pth升级也升级不了:
>pip install --upgrade --ignore-installed scikit-allel -i https://pypi.tuna.tsinghua.edu.cn/simple
.......
ERROR: Command errored out with exit status 1:没办法放弃conda安装,直接从GitHub安装:https://github.com/hardingnj/xpclr。
git clone https://github.com/hardingnj/xpclr.git
cd xpclr
python setup.py install进入bin后,可直接运行xpclr:
$ xpclr -h
usage: xpclr [-h] --out OUT [--format FORMAT] [--input INPUT] [--gdistkey GDISTKEY] [--samplesA SAMPLESA] [--samplesB SAMPLESB] [--rrate RRATE] [--map MAP] [--popA POPA] [--popB POPB]
--chr CHROM [--ld LDCUTOFF] [--phased] [--verbose VERBOSE] [--maxsnps MAXSNPS] [--minsnps MINSNPS] [--size SIZE] [--start START] [--stop STOP] [--step STEP]
Tool to calculate XP-CLR as per Chen, Patterson, Reich 2010需要注意的是,你如果使用xpclr的全路径是运行不了的:
$ /project/xpclr/bin/xpclr
Traceback (most recent call last):
File "/project/xpclr/bin/xpclr", line 4, in
import numpy as np
ImportError: No module named numpy安装依赖包:pip install -r requirements.txt -i https://pypi.tuna.tsinghua.edu.cn/simple。
这时,也可以用python(实际上我当前的版本已经是Python 3.9.1,所以xpclr也是支持的。)来调用全路径:
python /project/xpclr/bin/xpclr --format vcf --input /project/04.sweep/sample750_miss0.6_impute/meanDP3.miss0.6.maf0.01.impute.rename.vcf --samplesA /project/04.sweep/sample750_miss0.6_impute/List/w-l-c/Cultivar.list --samplesB /project/04.sweep/sample750_miss0.6_impute/List/w-l-c/Wild.list --chr 1 --maxsnps 600 --size 1000 --step 1000 --out test_out建议还是将安装路径/project/xpclr/bin加入环境变量(测试了下,貌似不用加入环境变量也可直接调用,可能是软件安装环节已经加入),直接用xpclr。
原因分析
conda安装虽然便捷,但作者没有及时更新,最早的版本也是在3年前(Python2),导致一些包不兼容。
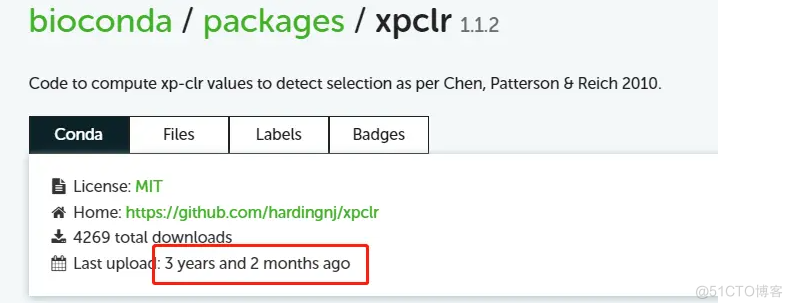
而GitHub版本作者还是在维护的,所以对于后来者推荐之。
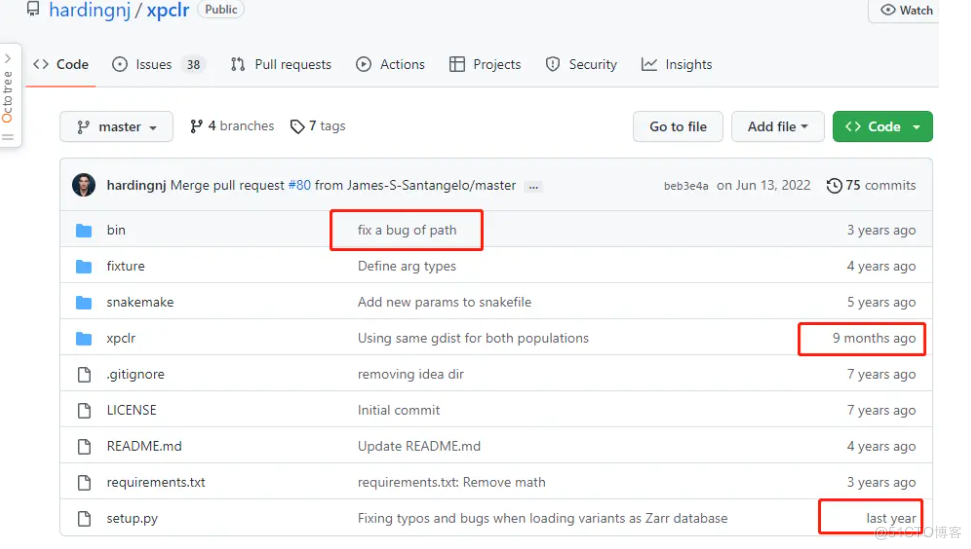
后续分析推荐
原版XP-CLR用法参考:https://zhuanlan.zhihu.com/p/145387269
Python版XP-CLR用法参考:https://www.jianshu.com/p/9c827a0be66d
作者:Bioinfarmer,微信公众号:Bioinfarmer。
服务器托管,北京服务器托管,服务器租用 http://www.fwqtg.net
机房租用,北京机房租用,IDC机房托管, http://www.fwqtg.net
