
Deepro-Glu: combination of convolutional neural network and Bi-LSTM models using ProtBert and handcrafted features to identify l
会议时间:2022-10-30
会议地点:腾讯会议
关键词:lysine glutaryation, BERT , deep le服务器托管网arning, protein language models
作者:Xiao Wang
期刊:Bioinformatics
年份:2022
论文原文:https://doi.org/10.1093/bib/bbac631代码:https://github.com/xwanggroup/Deepro-Glu
主要内容
1问题 :赖氨酸谷氨酰化(Kglu)是一种新发现的蛋白质翻译后修饰,在线粒体功能、氧化损伤等方面具有重要作用。确定谷氨酰化位点的既定生物实验方法往往耗时且成本高。因此,迫切需要开发计算方法来有效和准确地识别谷氨酰化位点。现有的大多数计算方法只利用手工制作的特征来构建预测模型,而没有考虑预训练的蛋白质语言模型对预测性能的积极影响。基于此,我们开发了一个集合的深度学习预测器Deepro-Glu,它结合了卷积神经网络和双向长短期记忆网络,利用深度学习特征和传统的手工特征预测赖氨酸谷氨酸化位点。深度学习特征是由预先训练好的蛋白质语言模型ProtBert生成的,手工制作的特征包括基于序列的特征、基于物理化学性质的特征和基于进化信息的特征。此外,注意力机制被用来通过学习适当的注意力权重有效地整合深度学习特征和手工制作的特征。10倍交叉验证和独立测试表明,Deepro-Glu取得了比最先进的方法更有竞争力或更高的性能。
2方法
在这项研究中,我们使用实验验证的赖氨酸谷氨酰化的基准数据集来评估所提出的Deepro-Glu模型的性能,其中所有的数据都来自于蛋白质的通用蛋白质(UniProt)数据库,由UniProt的高级搜索和注释功能创建。根据Chou的方案,含有潜在谷氨酰化位点的多肽样品一般可以表示为
(1)P.(K) R-ER-(E-1)-R-1KR+1 -………
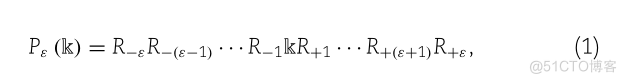
其中符号’k’表示蛋白质片段中的赖氨酸,下标是一个整数。R-代表来自中心的第个上游残基,R+代表第个下游氨基酸残基,以此类推。所以很明显,肽P(k)的长度是2+1。肽样本P(k)的(2+1)元组可以进一步划分为以下两类
PE(K)P.(K)EPE(K)

其中P+(k)表示以赖氨酸为中心的真实谷氨酸化段,P-(k)表示以赖氨酸为中心的虚假谷氨酸化肽。
此外,目前研究的基准数据集可以表示为
ANLY.

其中Zk是赖氨酸-谷氨酰胺化的基准数据集,正子集Z+k只包含赖氨酸-谷氨酰胺化的样本,负子集Z-k只包含非赖氨酸-谷氨酰胺化的样本。 最后一个符号∪是集合理论中的 “联合 “符号。
经过一些初步的测试,同时考虑到之前研究者的处理,我们选择了=20。据此,每个蛋白质片段被裂解到41的长度,所以总共得到2405个肽的样品,正样品:954,负样本:1451。由于赖氨酸 “k “需要在中心位置,如果切割后的肽没有达到指定的长度,我们将使用一个假的氨基酸 “X “来填充序列,以保持上游残基和下游残基的数量相同的大小。最后 ,为了验证模型,我们将基准数据集分为两部分,训练集:1683,测试集:722,其中正负样本的比例为4:6,并确保测试集的数据不出现在训练集中。
特征表示
在本小节中,我们将传统的手工制作的特征和从预训练的ProtBert模型中提取的特征结合起来,作为DNN的输入。那些传统的手工制作的特征根据蛋白质的主要结构、物理化学性质和进化信息分为三类。与以前的方法相比,其优势在于整合了各个层次的特征,可以从多个角度更全面地挖掘赖氨酸残基之间的隐含信息。
BE
二元编码是一种简单的编码方案,它描述了蛋白质序列中谷氨酰化位点和非谷氨酰化位点周围氨基酸的相对位置和类型。这种编码方案通过将原始基序字符信号转换为数字信号来生成输入预测器的特征。每个氨基酸残基可以表示为一个21维的二进制向量。例如,残基 “D “可以表示为(001000000000000000000),虚拟残基 “X “可以表示为(00000000000000000000001)。在这项研究中,一个窗口大小为41的肽序列可以被编码为41∗21=861维的特征。
DDE
2015 年,Saravanan 等人 [40]。提出了二肽偏离预期平均值(DDE)的概念。 DDE 特征向量由三个主要参数计算:二肽组成的测量、二肽的理论平均值和二肽的理论方差。上述三个参数和 DDE 的公式表示如下。
DEO 二 分.

其中 Cc(i) 表示肽段 P 上二肽 i 的二肽组成 Cc(i),共有 400 个二肽组合(2020 种常见氨基酸),ni 是二肽 i 的显示频率和N 为 l − 1(即肽段 P 中可能存在的二肽数量)。接下来,如下计算蛋白质序列的理论平均值(Tm)和理论方差(Tv)。
CIITM.CNCN
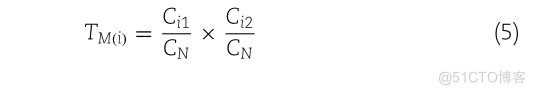
Ci1和Ci2分别是编码第一个氨基酸的密码子数和编码第二个氨基酸的密码子数,而CN是排除三个终止密码子的可能密码子总数。由于 TM(i) 不依赖于肽 P,因此已预先计算了 400 个二肽。
TMGI (1 – TMGI)TVON
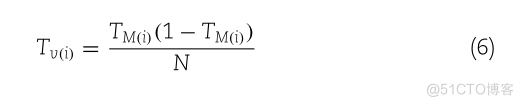
DDE 特征向量基于 Tm、Tv 和 Dc。最后,DDE(i) 指定为
DE(I)TMCI)DDE(T)VO)
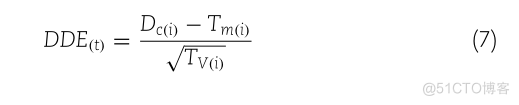
对于DDE函数得分矩阵,计算出的400维特征向量表示如下:
DDEPDDE,DDE@],I,1,2,…….400
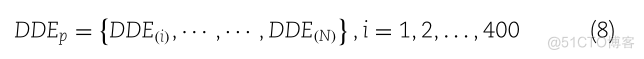
AAindex
AAindex 氨基酸指数数据库(AAindex)是一个数字指数数据库,它包括氨基酸残基和其他形式的蛋白质序列的各种物理、生物和化学特性。
在本研究中,我们选取了12种类型
TABLE 1.THE SELECTED AAINDEX PROPERTIESNO.AAINDEX IDNO.AAINDEX ID71TSAJ990101NOZY71010128MAXF760101KLEP84010139NAKH900109NAKH920108104LIFS790101BLAM930101511HUTI700103BIOV880101612CEDF970104MIYS990104

因此,基于AAindex的特征向量的维数为41*12=492。
BLOSUM62
BLOSUM矩阵是一个常用的氨基酸替换评分矩阵,评估进化不同的蛋白质序列之间的分数。它是基于氨基酸序列的比较,两个肽序列之间的同源性不超过62%。在这里,我们用BLOSUM62来转换蛋白质的主序列,以显示序列的两个片段之间的相似性。最终,基于BLOSUM62的特征向量的维度为41*20=820。
模型
MODEL_1RMITKIRMULTI-HEAD ATTENTIONAVERAGE POOLINGFEED FORWARDADD&NORMADD&NORMDATTENTIONPROTBERTCONYOLUTIONSEQMODELRM TKIRBEAVERAGE POOLINGBI-LSTMDDEOUTPUTAAINDEX0NBLOSUM62..DMLPFEATURE EXTRACTBI-LSTMSEQ
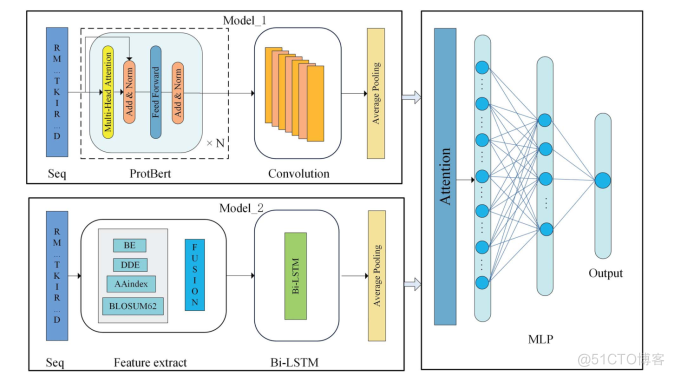
提出的框架Deepro-Glu的概述。在Model_1中,原始蛋白质序列被送入预先训练好的ProtBert语言模型以学习有效的蛋白质表征,然后使用1D-CNN来捕获重要的蛋白质语义信息。在Model_2中,融合后的手工特征被送入Bi-LSTM以学习序列信息。两个模型的平均池化层用于降低特征的维度并去除冗余信息。之后,这两个模型被组合起来,并被送入注意力机制以学习关键信息。最后,通过多层感知器得到预测结果。
预先训练的ProtBert语言模型
BERT的模型结构是一个典型的双向编码模型,使用多层双向Transformer Encoder块进行连接。每个编码器模块由一个多头的自我注意子层和一个前馈神经网络子层组成;残差连接被部署在这些子层周围,然后进行层的规范化。
微调后的BERT模型改进了各种蛋白质预测任务,并将新模型命名为ProtBert。ProtBert模型在包含2亿多条蛋白质序列的BDF和UniRef数据库中进行了预训练。该模型将原始模型的层数增加到30层,并以自我监督的方式预训练了大量的原始蛋白质序列。
这是一个自监督的蛋白质语言模型,该模型已成功应用于不同的下游任务,这也增加了我们使用从ProtBert提取的特征的信心。
具体来说 ,ProtBert模型的输入是原始蛋白质序列,本研究中设置的蛋白质窗口大小为41,因此预训练模型的向量维数为102 441。在本研究中,从预训练的语言模型得到的蛋白质表征被降维。也就是说,在网络结构中加入了一个平均池层,这样做的好处是减少了冗余特征对最终预测精度的影响。然后,这些语言模型特征将与三种手工制作的特征相结合。而这些融合后的特征将被送入注意力机制,最终得到赖氨酸谷氨酰胺化位点的预测结果。
3主要实验及结果
氨基酸位置分布的分析
为了说明赖氨酸谷氨酰化位点两侧残基的不同分布和偏好,比较了观察到的Glu位点周围的氨基酸序列和非Glu位点的氨基酸序列。
GLUTARYLATION AND NON-GLUTARYLATION PEPTIDES48.4%LPTSYD荷花AKGENRICHEDCLESHHAQGVRAGETAPVTVEHRRAMGTVP早TTLAINIGKDEVALE店BHDPMAFHLPOFQVLKSSQ!GPDEPLETEDSVEKELRRRRAIG4R 4%FIGURE 2.MOTIF CONSERVATION ANALYSIS OF GLU SITES USING THE TWO-SAMPLE-LOGOS (T-TEST WITH P

谷氨酰化和非谷氨酰化序列之间以及上游(位置-20至-1)和下游段(位置1-20)之间都有明显的差异。氨基酸残基K, G明显存在于耗竭区的上游,而T , V明显存在于富集区的上游。对于下游区域,残基R, L明显出现在负样本一侧,而P , E明显出现在正样本一侧。总之,赖氨酸周围的氨基酸残基的频率存在明显差异,这为利用序列信息进行建模计算分类以预测谷氨酰化位点提供了前提条件。
几种手工制作的特征编码方法的性能评估
在10倍交叉验证的基础上,使用Bi-LSTM对四种不同的单一特征和它们的融合特征的性能进行了评估
10.950.90.850.服务器托管网80.750.70.650.60.550.5SPMCCSNPREACCFUSION FEATURESDDEBEBLOSUM62AAINDEXFIGURE 3.COMPARISON OF THE PERFORMANCE ON DIFFERENT HANDCRAFTED FEATURES BASED ON TRAINININING SE
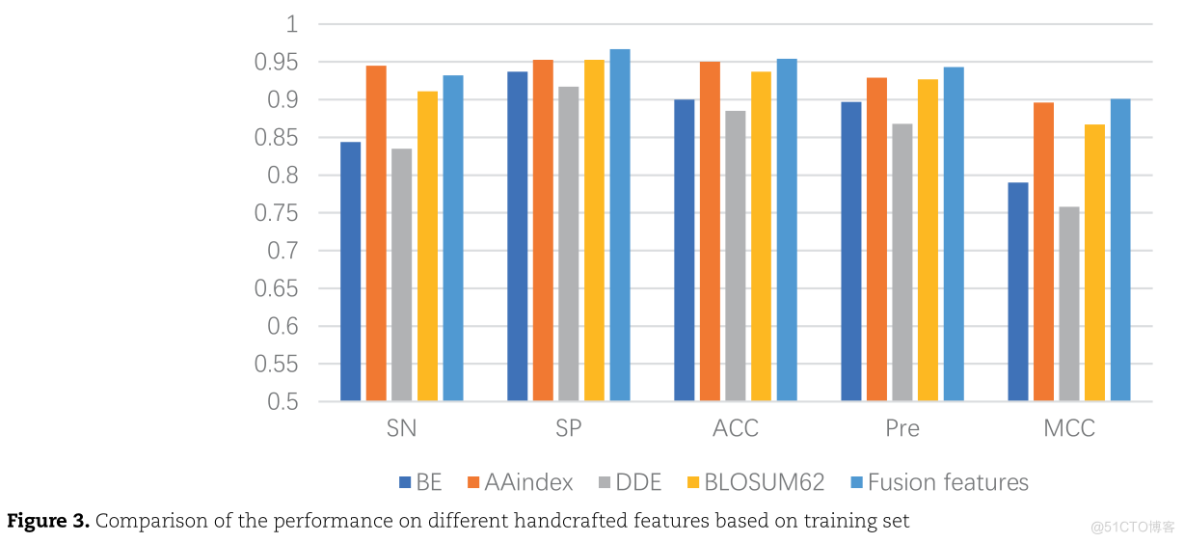
BE和DDE特征是基于序列信息的,AAindex特征是基于物理化学性质的,BLOSUM62矩阵是基于进化信息的。
在单个特征中,AAindex特征的性能结果最好,然而,在整合这四种特征表示后,总体准确率提高了0.4-6.9%。这说明单一的特征提取方法有一定的局限性,而融合特征表示有利于从各个方面表示蛋白质序列,从而提高模型的预测性能。
模型参数优化
为了探索DeeproGlu预测器的最佳参数组合,我们根据模型的10倍交叉验证的平均结果来选择参数。设置了不同的卷积单元和卷积核来进行实验。表2显示了不同CNN参数在训练集上的性能比较。
TABLE 2. PERFORMANCE COMPARISON OF DIFFERENT CNN PARAMETERS ON TRAINING SEUNITS128UNITS8UNITS32UNITS64UNITS16KERNEL SIZEMCCMCCMCCMCCMCC0.9080.9060.9230.9150.9100.8970.9130.9150.8970.9060.8880.9170.9030.8780.914
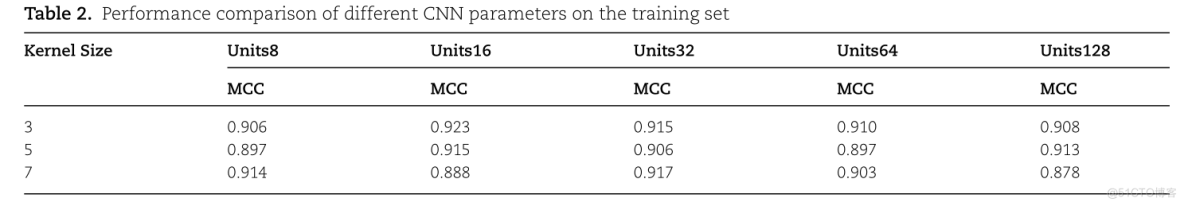
Kernel Size表示卷积层中卷积核的大小。我们将卷积核的大小设置为3、5和7,以找到模型的最佳卷积核大小。此外,Units8、Units16、Units32、Units64和Units128分别表示卷积层中卷积单元的数量为8、16、32、64和128。从T able 2可以看出,当卷积核为3,卷积单元的大小为16时,模型的性能是最好的。
不同CNN层下的模型性能
TABLE 3. PERFORMANCE COMPARISON OF DIFFERENT CNN STRUCTURE MODELS ON TRAINININININININING SETSNACCSPSPMODELAUPRPRE0.9700.9540.9231LAYER0.9540.9630.9872LAYER0.9150.9670.8880.9420.9480.9613LAYER0.9210.9720.9740.9190.9610.948
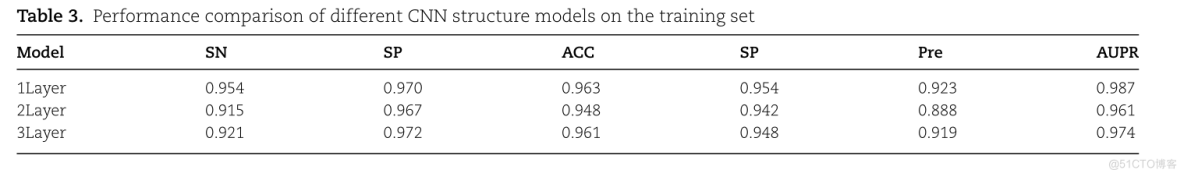
单层CNN结构的模型在训练集上取得了最好的预测性能;SN、Pre、MCC和精度-召回曲线下面积(AUPR)的指标值为3.3-3.9%,0.6-1.2%。比其他两个CNN层数不同的模型分别高0.4-3.5%和1.3-2.6%。
不同LSTM结构下的模型性能,包括单向LSTM和双向LSTM以及LSTM层的数量。
TABLE 4. PERFORMANCE COMPARISON OF DIFFERENT LSTM STRUCTURE MODELS ON THE TRAINININING SETMODELACCAUPRSPMCCSNPRE1LAYERF0.9740.9380.9020.9640.9540.93821AYERF0.9100.9870.9670.9110.9580.9760.9780.8971LAYERT0.9700.9210.9520.9480.9230.9870.9630.9540.95421AYERT0.9700.9100.9440.9440.9583LAYERT0.9780.967
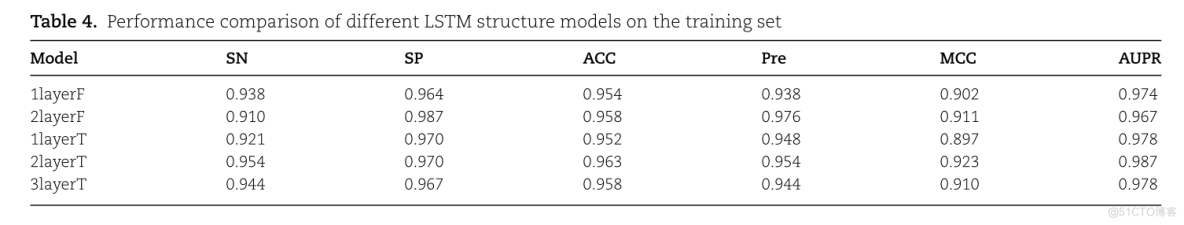
1layerF表示该模型为单层单向LSTM
1layerT表示该模型为单层双向LSTM
如图4所示,单层双向LSTM模型结构比单层单向LSTM模型结构好。
我们分析其原因是单方向LSTM状态的传递是从前到后单向的,限制了信息传播的方向,而双向LSTM可以同时学习前向和后向信息,增加了神经网络的表现力。至于Bi-LSTM模型的层数,从Table 4可以看出,两层Bi-LSTM模型的性能最好,在SN、Pre、MCC和AUPR方面分别比三层模型高1%、1.1%、1.3%和0.9%。
选择Deepro-Glu的模型架构
我们通过分析使用预训练的语言模型和序列(SC)特征与不同的深度学习特征提取器相结合的训练数据来比较多种模型架构。为了研究模型中提出的一些结构是否有效,我们将去除结构的网络得到的结果与加入结构的网络得到的结果进行了比较。
TABLE 5. 10-FOLD CROSS-VALIDATION RESULTS OF DIFFERENT MODELS ON THE TRAINININING SETSPAUCSNPREMCCACCMODEL0.8490.9250.911SC-CNN0.8590.7680.8920.9760.8790.9360.9100.9440.964SC-LSTM0.943SC-BILSTM0.9780.9670.9320.9010.954SC-BILSTM+PROTBERT-CNN0.9320.9770.9850.9590.9600.9150.9540.9880.9630.954DEEPRO-GLU0.9230.970
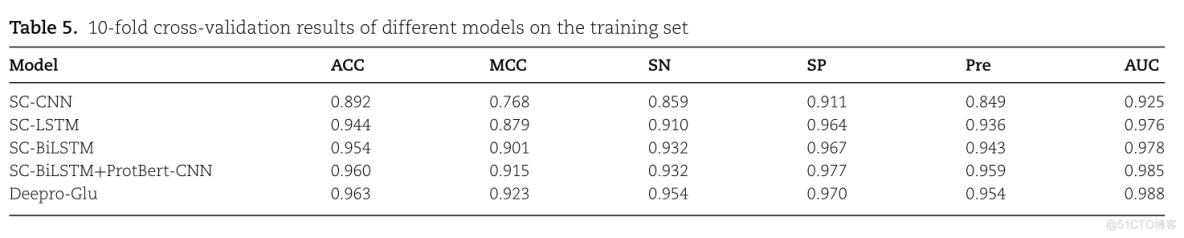
手工制作的特征与LSTM网络相结合的总体性能不如与Bi-LSTM网络相结合的性能。我们分析一下Bi-LSTM优于LSTM的原因:双向LSTM网络可以学习前向和后向序列之间的长程依赖关系,这可以帮助更好地理解序列上下文。从Table 5可以看出,与Bi-LSTM结合的生物序列单个模型的灵敏度、特异性、准确性、Mathews相关系数和精度分别为0.932、0.967、0.954、0.901和0.943,而集合深度学习模型的指标高于所有单个模型,这充分证明了集合深度学习模型的有效性。
在这项研究中,我们还比较了训练集上带有和不带有注意力机制的模型的结果。没有注意机制的模型的准确度为0.960,MCC为0.915;而有注意机制的模型的准确度为0.963,MCC为0.923。MCC是对模型整体性能的衡量,在不平衡数据集的条件下,MCC是比准确率更好的性能评价指标。因此,基于准确率和MCC都优于没有注意机制的模型,我们可以证明注意机制在谷氨酰化位点的预测中起着重要作用。
精度-召回曲线见图4
PRECISION-RECALL CURVES1.00.8PRECISION0.60.4SC-CNN(AREA0.873)二二二SC-LSTM(AREA0.975)SC-BILSTM(AREA0.977)0.2SC-BILSTM+PROTBERT-CNN(AREA0.981)DEEPRO-GLU(AREA0.987)0.00.40.21.00.60.80.0RECALLFIGURE 4. PRECISION-RECALL CURVES WITH DIFFERENT MODELS ON THE TRAIN-ING SET.

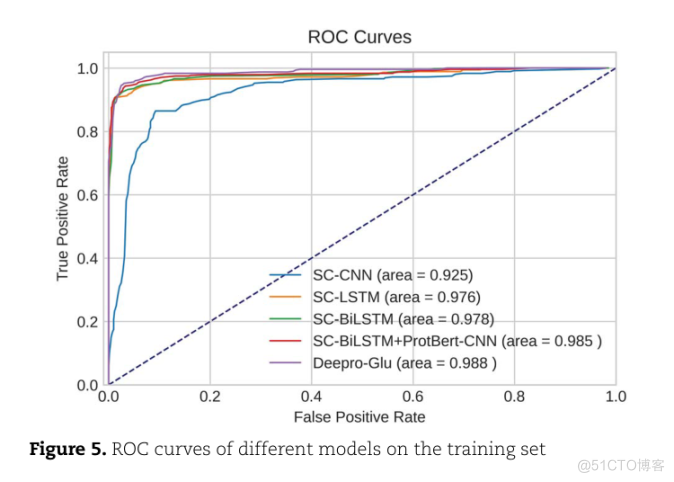
ROC曲线见图5
ROC CURVES1.00.8TRUE POSITIVE RATE0.60.4SC-CNN(AREA0.925)SC-LSTM(AREA0.976)SC-BILSTM(AREA0.978)0.2SC-BILSTM+PROTBERT-CNN(AREA985)DEEPRO-GLU(AREA00.988)0.00.40.21.00.60.00.8FALSE POSITIVE RATEFIGURE 5.ROC CURVES OF DIFFERENT MODELS OFMODELS ON THE TRAINING SET
与现有预测器的比较
由于预测器GlutPred、MDDGlutar的测试网站目前无法访问,DeeproGlu只与iGlu-Lys、DeepGlut、iGluK-Deep和DNN-E进行比较
文献显示,在类不平衡问题上,MCC是比准确率和F1得分更好的性能评估指标。较高的MCC值表明所提出的方法在预测赖氨酸谷氨酰化位点方面有更好的表现。此外,DeeproGlu也是第一个在赖氨酸谷氨酰化位点预测问题上达到90%的MCC值的预测器。
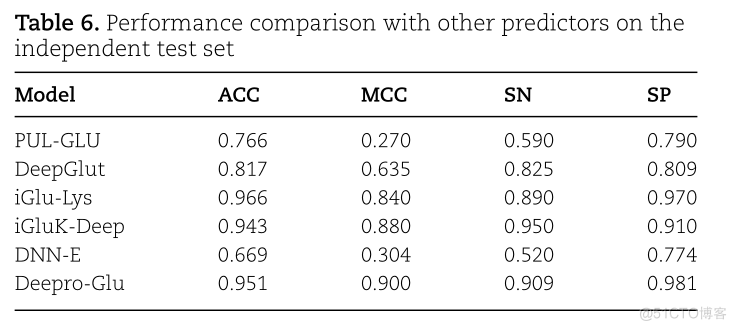
TABLE 6.PERFORMANCE COMPARISON WITH OTHER PREDICTORS ON THEINDEPENDENT TEST SETMODEISNMCCSPACC0.7900.7660.2700.590PUL-GLUDEEPGLUT0.8090.8170.8250.6350.9700.966IGLU-LYS0.8400.890IGLUK-DEEP0.9430.9100.9500.8800.7740.3040.520DNN-E0.6690.9810.9510.9000.909DEEPRO-GLU
而该模型的混淆矩阵如图6所示。
CONFUSION MATRIX ON INDEPENDENT TEST SET4003508414NON-GLU300250TRUE LABEL20015027271PLCO10050NON-GLUGLUPREDICTED LABEL
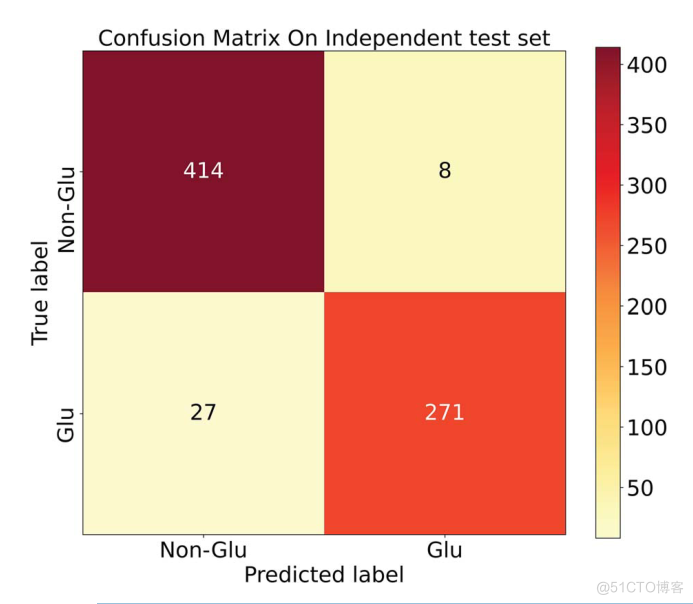
Deepro-Glu对独立测试集的混淆矩阵。Glu表示谷氨酰化位点为正,Non-Glu表示谷氨酰化位点为负。
4结论
本研究提出了一个名为Deepro-Glu的预测器,它使用一个集合深度学习框架来预测赖氨酸谷氨酰胺化位点。本文的主要贡献有以下几点。(1) 从更全面的角度表现了蛋白质的特征:从预训练的ProtBert模型中提取的特征和传统的手工制作的特征三种不同的类型,这些手工制作的特征分别基于蛋白质主序列、蛋白质理化性质和蛋白质进化信息。 (2) 使用集合深度学习架构来构建我们的预测模型。第一个子模型使用1D-CNN来捕捉ProtBert模型中的蛋白质信息,第二个子模型使用Bi-LSTM来学习手工特征中的序列信息。(3)利用注意力机制,从融合特征中有效地捕捉关键信息。
服务器托管,北京服务器托管,服务器租用 http://www.fwqtg.net
机房租用,北京机房租用,IDC机房托管, http://www.fwqtg.net
执行性思维:人工智能的现实优势 如何解构人类的思维模型是一个跨多学科的综合性问题。本文仅针对AI领域发展方向预测以及理解,提出一个简化的模型。我认为人类的思维基于思考的目的性可以分为:执行性思维和创造性思维两种 定义:基于既定模型和规则的计算性思维 执行性思维…
