
https://wilkox.org/gggenes/gggenes是ggplot2的扩展包,用于绘制基因结构图、多物种基因比较图的很好玩的工具。
- 1初识ggplot2绘制几何对象
- 12个ggplot2扩展包帮你实现更强大的可视化
- ggplot2学习笔记之图形排列
- ggplot2高效实用指南 (可视化脚本、工具、套路、配色)
- 一个震撼的交互型3D可视化R包 – 可直接转ggplot2图为3D

安装
一种是安装稳定版本的gggene
install.packages("gggenes")另一种是从github上安装开发版
devtools::install_github("wilkox/gggenes")下面是用的数据内容如下:
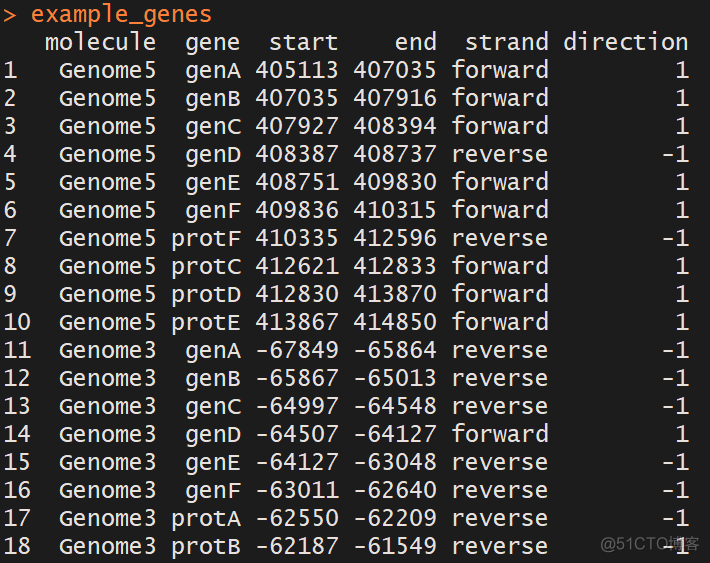
example_genes包括118行和6个变量。每一行代表一个基因或一个区域;列分别是:
- molecule:基因组名字
- gene: 基因名字 the name of the gene
- start: 基因在基因组开始位置 (如果在负链,注意起始位置的写法跟
bed文件不同了) - end: 基因结束位置 (负链的基因起始位置绝对值大于结束位置)
- strand: 基因属于哪条链 (可选)
如果想显示基因的子区域,如外显子、或翻译为特定功能域的区域等。
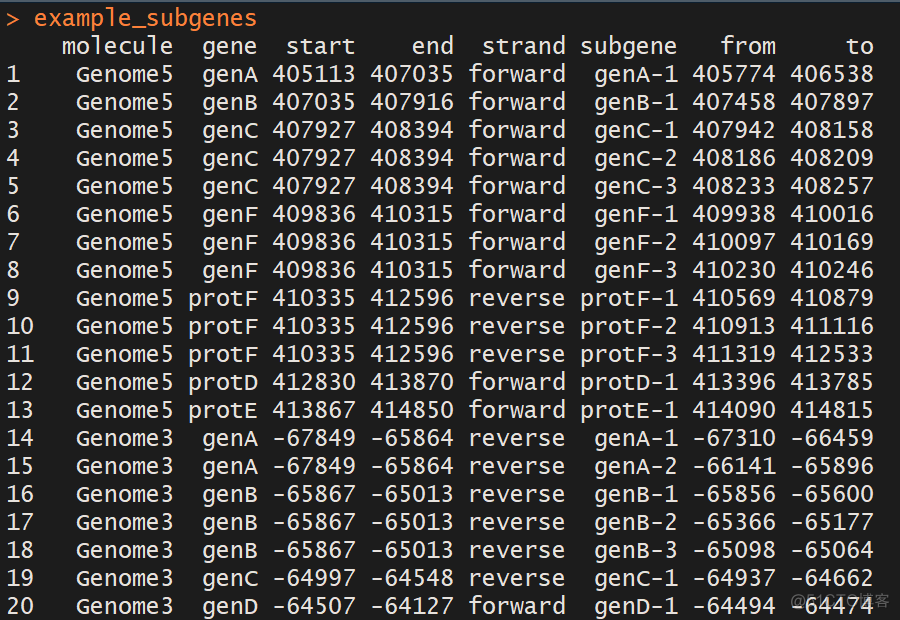
example_subgenes多了三列信息:
- subgeme: 子片段名字
- from: 子片段开始位置
- to: 子片段结束位置
用geom_gene_arrow()画基因箭头
geom_gene_arrow()是一个ggplot2几何性状,它用箭头表示基因。基因在分子内的起始和结束位置分别映射到xmin和xmax。这些开始和结束位置用于确定箭头指向的方向。基因组信息molecule映射到y轴。如果绘制的基因来自不同基因组的位置的数值相差很大,一般指定scale =“free”来调整横轴的坐标展示,以避免部分数字太大压缩了小基因组的基因的展示。
library(ggplot2)
library(gggenes)
ggplot(example_genes, aes(xmin = start, xmax = end, y = molecule, fill = gene)) +
geom_gene_arrow() +
facet_wrap(~ molecule, scales = "free", ncol = 1) +
scale_fill_brewer(palette = "Set3")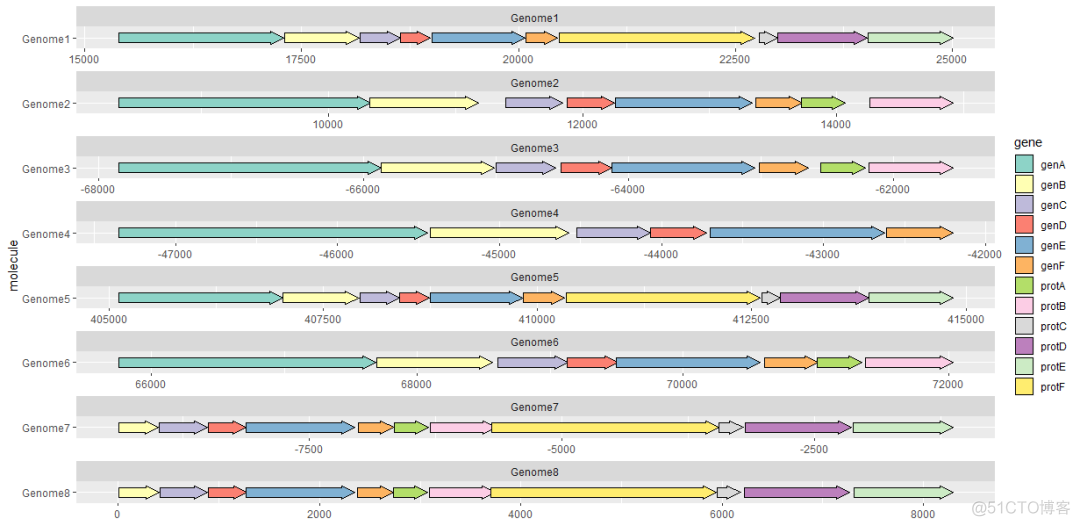
用theme_genes美化图形
由于生成的图可能看起来很混乱,因此ggplot2主题theme_genes提供了一些合理的缺省值美化结果。
ggplot(example_genes, aes(xmin = start, xmax = end, y = molecule, fill = gene)) +
geom_gene_arrow() +
facet_wrap(~ molecule, scales = "free", ncol = 1) +
scale_fill_brewer(palette = "Set3") +
theme_genes()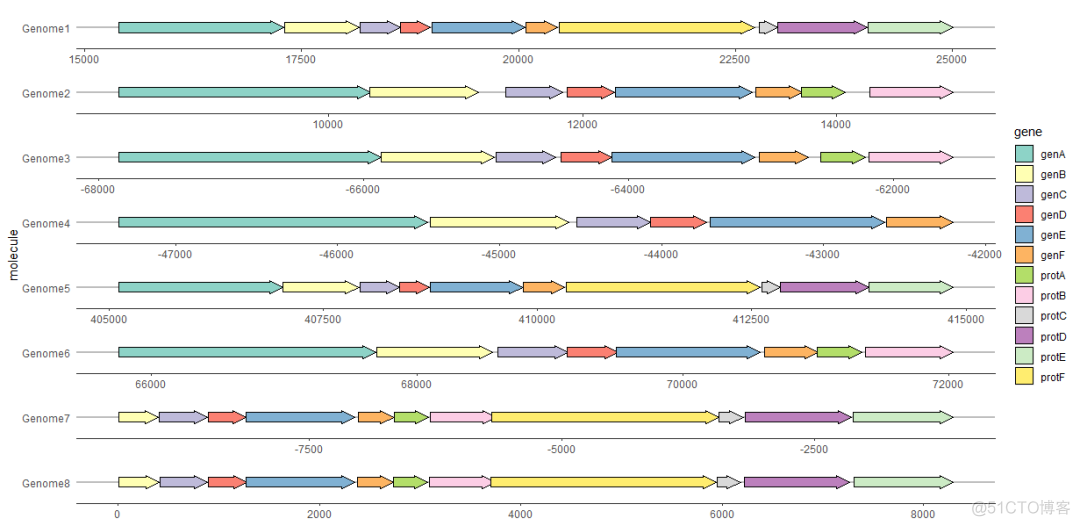
使用make_alignment_dummies()跨面对齐基因
通常我们会想要所有物种按某一个指定的基因对齐,比如下面例子中的geneE。make_alignment_dummies()会根据给定的数据和待对齐的基因,生成一组空基因;再使用geom_blank()将这些空基因添加到绘图中,就可以填充两侧的空白,以在图上直观地对齐所选的基因。
dummies 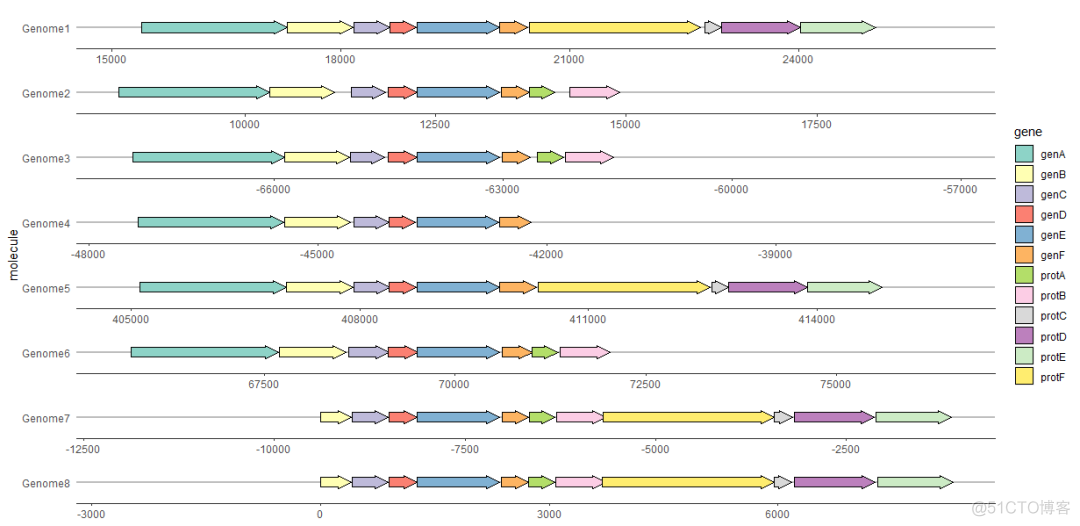
用geom_gene_label()标记基因
把基因名字所在的列名字映射到label属性可以在图上标记每个基因的名字。geom_gene_label()使用ggfittext包将标签文本放入基因箭头内。
ggplot(
example_genes,
aes(xmin = start, xmax = end, y = molecule, fill = gene, label = gene)
) +
geom_gene_arrow(arrowhead_height = unit(3, "mm"), arrowhead_width = unit(1, "mm")) +
geom_gene_label(align = "left") +
geom_blank(data = dummies) +
facet_wrap(~ molecule, scales = "free", ncol = 1) +
scale_fill_brewer(palette = "Set3") +
theme_genes()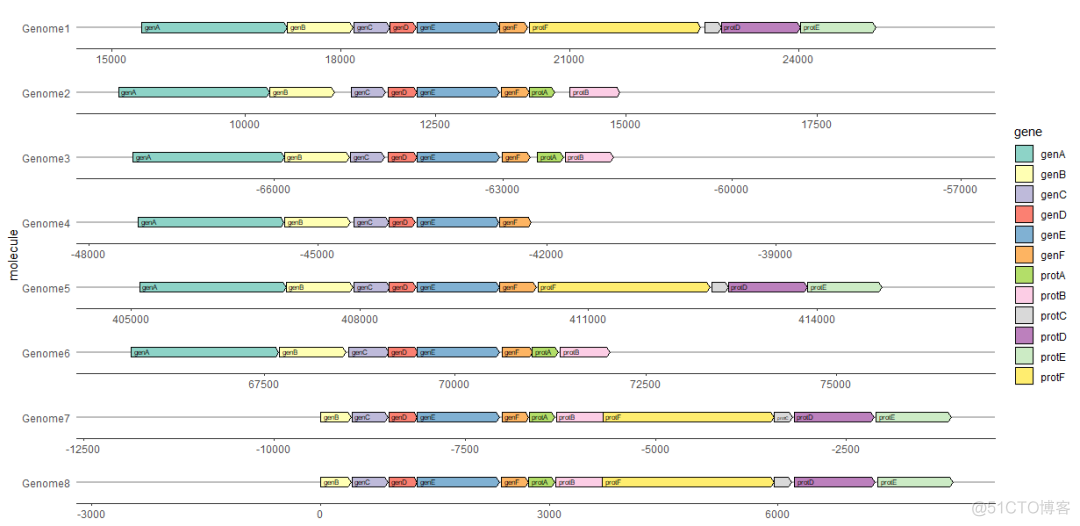
正负链基因分开展示
forward属性可以用于在同一张图分开正负链基因的展示。如果forward为TRUE(默认值),或者任何强制为TRUE的值(如1),则该基因将被绘制为指向正常方向,即xmin和xmax所暗指的方向。如果forward为FALSE,或者任何强制为假的值(如-1),则该基因将按暗指方向的相反方向绘制。
在下面的例子中,forward被用来反转所有反链上所有的基因方向,与xmin和xmax暗指的方向相反。
example_genes$direction 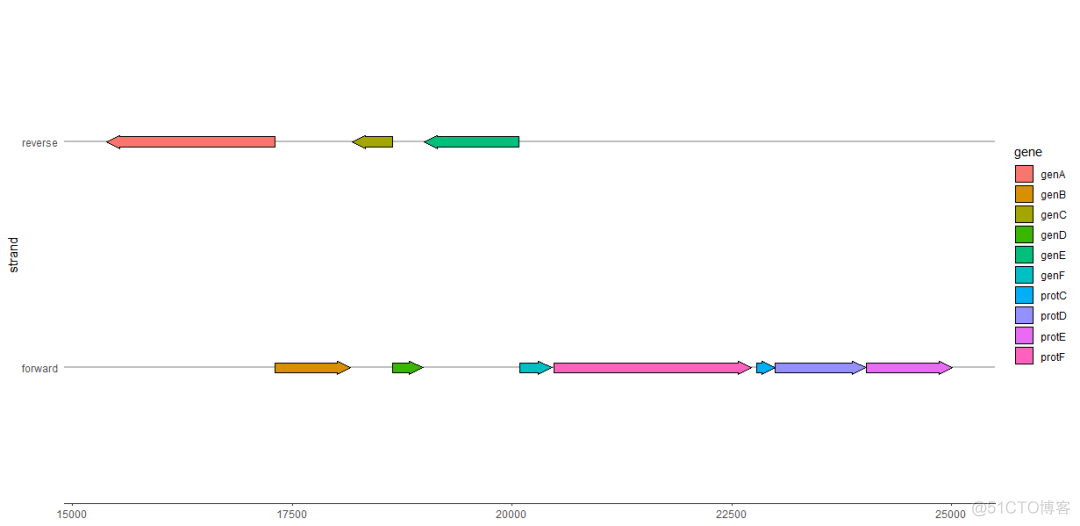
查看子基因(subgene)片段
我们可以使用geom_subgene_arrow()突出显示子基因片段,例如蛋白功能域或局部比对区域。
这与geom_gene_arrow()类似,但是除了xmin和xmax(确定基因边界)之外,我们还需要xsubmin和xsubmax来确定子区域的边界。如果子区域的边界超出了基因区域,则跳过该子区域,并弹出警告。配合geom_gene_arrow()不给基因上色,而只标记子区域。
ggplot(example_genes, aes(xmin = start, xmax = end, y = molecule)) +
facet_wrap(~ molecule, scales = "free", ncol = 1) +
geom_gene_arrow(fill = "white") +
geom_subgene_arrow(data = example_subgenes,
aes(xmin = start, xmax = end, y = molecule, fill = gene,
xsubmin = from, xsubmax = to), color="black", alpha=.7) +
theme_genes()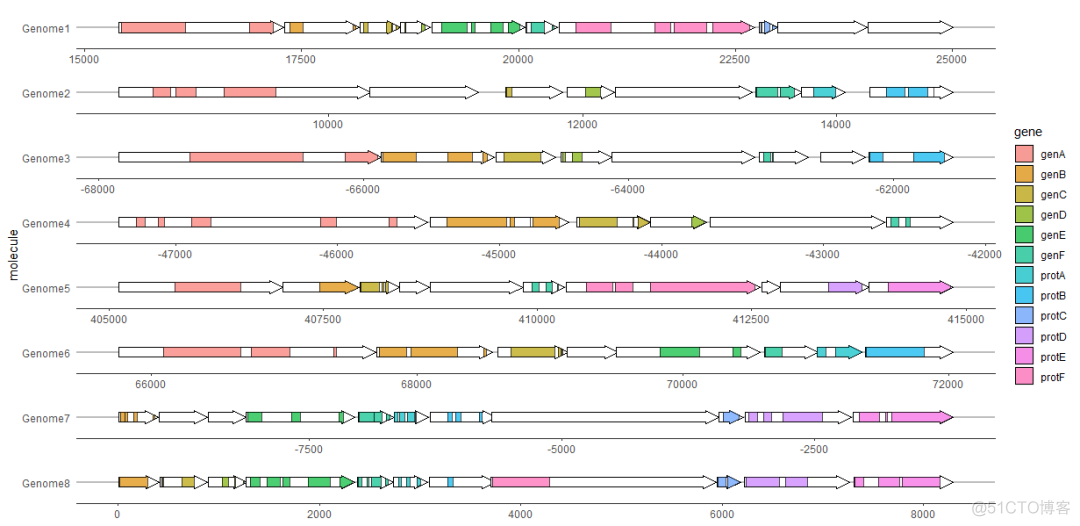
使用geom_subgene_label()给子区域在图上加标签,它的工作原理类似于geom_gene_label(),但主要的区别是它需要xsubmin和xsubmax属性 (而不是xmin和xmax)。
ggplot(subset(example_genes, molecule == "Genome4" & gene == "genA"),
aes(xmin = start, xmax = end, y = strand)
) +
geom_gene_arrow() +
geom_gene_label(aes(label = gene)) +
geom_subgene_arrow(
data = subset(example_subgenes, molecule == "Genome4" & gene == "genA"),
aes(xsubmin = from, xsubmax = to, fill = subgene)
) +
geom_subgene_label(
data = subset(example_subgenes, molecule == "Genome4" & gene == "genA"),
aes(xsubmin = from, xsubmax = to, label = subgene),
min.size = 0
)
高颜值免费在线绘图
R统计和作图
- 文章用图的修改和排版 (1)
- 文章用图的修改和排版 (2)
- 简单强大的在线绘图
- 简单强大的在线绘图-升级版
- 简单强大的在线绘图-第3版
- 论文图表基本规范
- 学术图表的基本配色方法
- 数据可视化基本套路总结
- Graphpad,经典绘图工具初学初探
- 你的包佩奇了吗?试试新版Rstudio,自动提醒缺失包!
- 原来Rstudio还可以这么使用,又方便了一些
- 在R中赞扬下努力工作的你,奖励一份CheatShet
- 别人的电子书,你的电子书,都在bookdown
- R语言 – 入门环境Rstudio
- R语言 – 热图绘制 (heatmap)
- R语言 – 基础概念和矩阵操作
- R语言 – 热图简化
- R语言 – 热图美化
- R语言 – 线图绘制
- R语言 – 线图一步法
- R语言 – 箱线图(小提琴图、抖动图、区域散点图)
- R语言 – 箱线图一步法
- R语言 – 火山图
- R语言 – 富集分析泡泡图
- R语言 – 散点图绘制
- R语言 – 韦恩图
- R语言 – 柱状图
- R语言 – 图形设置中英字体
- R语言 – 非参数法生存分析
- R语言 – 绘制seq logo图
- WGCNA分析,简单全面的最新教程
- psych +igraph:共表达网络构建
- 一文学会网络分析——Co-occurrence网络图在R中的实现
- 一文看懂PCA主成分分析
- 富集分析DotPlot,可以服
- 基因共表达聚类分析和可视化
- R中1010个热图绘制方法
- 还在用PCA降维?快学学大牛最爱的t-SNE算法吧, 附Python/R代码
- 一个函数抓取代谢组学权威数据库HMDB的所有表格数据
- 文章用图的修改和排版
- network3D: 交互式桑基图
- network3D 交互式网络生成
- Seq logo 在线绘制工具——Weblogo
- 生物AI插图素材获取和拼装指导
- ggplot2高效实用指南 (可视化脚本、工具、套路、配色)
- 图像处理R包magick学习笔记
- SOM基因表达聚类分析初探
- 利用gganimate可视化全球范围R-Ladies(R社区性别多样性组织)发展情况
- 一分钟绘制磷脂双分子层:AI零基础入门和基本图形绘制
- AI科研绘图(二):模式图的基本画法
- 你知道R中的赋值符号箭头(
- R语言可视化学习笔记之ggridges包
- 利用ComplexHeatmap绘制热图(一)
- ggplot2学习笔记之图形排列
- R包reshape2,轻松实现长、宽数据表格转换
- 用R在地图上绘制网络图的三种方法
- PCA主成分分析实战和可视化 附R代码和测试数据
- iTOL快速绘制颜值最高的进化树!
- 12个ggplot2扩展包帮你实现更强大的可视化
- 编程模板-R语言脚本写作:最简单的统计与绘图,包安装、命令行参数解析、文件读取、表格和矢量图输出
- R语言统计入门课程推荐——生物科学中的数据分析Data Analysis for the Life Sciences
- 数据可视化基本套路总结
- 你知道R中的赋值符号箭头
- 使用dplyr进行数据操作30例
- 交集intersect、并集union、找不同setdiff
- 1数据类型(向量、数组、矩阵、 列表和数据框)
- 2读写数据所需的主要函数、与外部环境交互
- 3数据筛选——提取对象的子集
- 4向量、矩阵的数学运算
- 5控制结构
- 6函数及作用域
- 7认识循环函数lapply和sapply
- 8分解数据框split和查看对象str
- 9模拟—随机数、抽样、线性模型
- 1初识ggplot2绘制几何对象
- 2图层的使用—基础、加标签、注释
- 3工具箱—误差线、加权数、展示数据分布
- 4语法基础
- 5通过图层构建图像
- 6标度、轴和图例
- 7定位-分面和坐标系
- 8主题设置、存储导出
- 9绘图需要的数据整理技术
- 创建属于自己的调色板
- 28个实用绘图包,总有几个适合你
- 热图绘制
- R做线性回归
- 绘图相关系数矩阵corrplot
- 相关矩阵可视化ggcorrplot
- 绘制交互式图形recharts
- 交互式可视化CanvasXpress
- 聚类分析factoextra
- LDA分析、作图及添加置信-ggord
- 解决散点图样品标签重叠ggrepel
- 添加P值或显著性标记ggpubr
- Alpha多样性稀释曲线rarefraction curve
- 堆叠柱状图各成分连线画法:突出组间变化
- 冲击图展示组间时间序列变化ggalluvial
- 桑基图riverplot
- 微生物环境因子分析ggvegan
- 五彩进化树与热图更配ggtree
- 多元回归树分析mvpart
- 随机森林randomForest 分类Classification 回归Regression
- 加权基因共表达网络分析WGCNA
- circlize包绘制circos-plot
- R语言搭建炫酷的线上博客系统
- 维恩(Venn)图绘制工具大全 (在线+R包)
- R包circlize:柱状图用腻了?试试好看的弦状图
- 获取pheatmap聚类后和标准化后的结果
- 增强火山图,要不要试一下?
- 一个震撼的交互型3D可视化R包 – 可直接转ggplot2图为3D
- 赠你一只金色的眼 – 富集分析和表达数据可视化
- 是Excel的图,不!是R的图
- 道友,来Rstudio里面看动画了
- 用了这么多年的PCA可视化竟然是错的!!!
- 万能转换:R图和统计表转成发表级的Word、PPT、Excel、HTML、Latex、矢量图等
- 那天空飘过的梅花月饼,是今年中秋最好的礼物
- 2019年诺贝尔生理医学奖揭晓 |动图展示历年生理学奖
- 2019年诺贝尔化学奖揭晓 |八一八,那些年的诺贝尔化学奖
- cellassign:用于肿瘤微环境分析的单细胞注释工具(9月Nature)
- maftools|TCGA肿瘤突变数据的汇总,分析和可视化
- ggThemeAssist|鼠标调整主题,并返回代码
服务器托管,北京服务器托管,服务器租用 http://www.fwqtg.net
机房租用,北京机房租用,IDC机房托管, http://www.e1idc.net
