
关键词:
作者:
期刊:IEEE Journal of Biomedical and Health Informatics
年份:2023
论文原文: 服务器托管网https://doi.org/10.1101/2022.09.02.506180
主要内容
1问题
:长链非编码RNA(LncRNAs)在调控基因表达和其他生物过程中起着至关重要的作用。
区分lncRNA和蛋白质编码转录本(PCTs)有助于研究人员深入了解lncRNA的形成机制及其与各种疾病相关的下游调控。
该工作允许不依赖先前生物知识的分类。具有几个主要特点:
整个LncDLSM由两部分组成,第一部分是基于分层输入神经网络的Hinn分析器,该分析器用于提取k-mer频率特征的高级特征。另一个部分是基于CNN的检测器,旨在提取光谱特征的高级特征。然后,我们使用另一个基于神经网络的预测模块对这些高层特征进行合并,最终识别出LncRNA。
采用了迁移学习方法来解决物种差异,通过微调人类模型来训练其他物种的模型。
LncDLSM在5个物种数据上取得了较好的灵敏度和特异度。
2方法
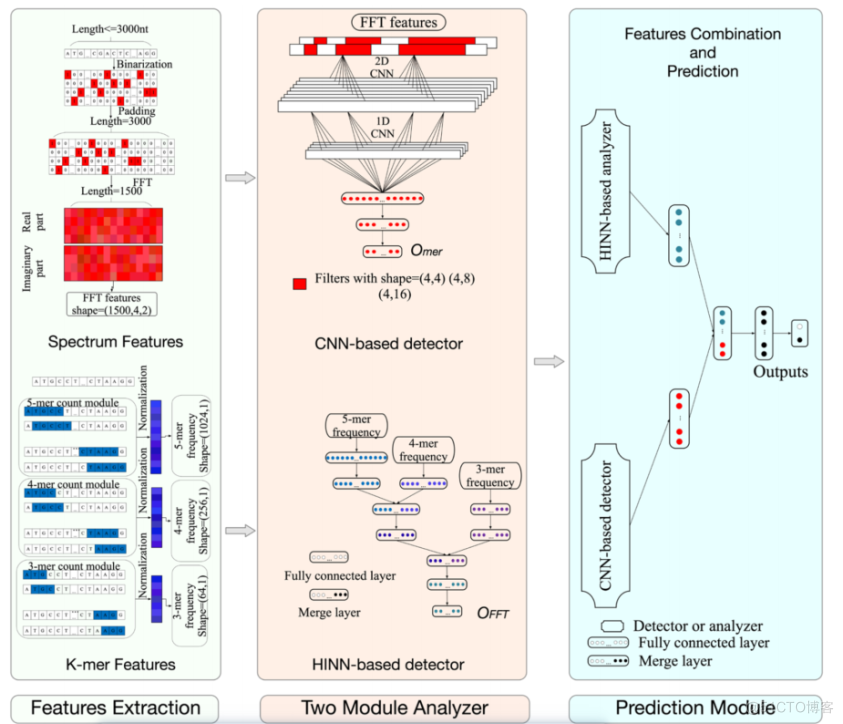
数据集
在NONCODE数据库中,人、小鼠、猪、牛和大鼠的LncRNAs数量分别为172216、131697、29585、23515和24879。
如图1所示,长度超过3000个核苷酸的lncRNAs仅覆盖约11.64%的人类数据和约10.30%的小鼠数据。
FIG.1.LENGTHDISTRIBUTIONOFTHEPCTSANDINCRNASONTHE(A)HUMANAND(B)MOUSEDATA10002000300040005000600SEGUENCELENGTHSEGUENCELENGTH200030004000.00030.000850006000INCRNAS0.0006.00100.00020.00010.00000.00040.00020.0007INCRNA0.0004PCTS0.0005.0000PCTS0.000610001000200
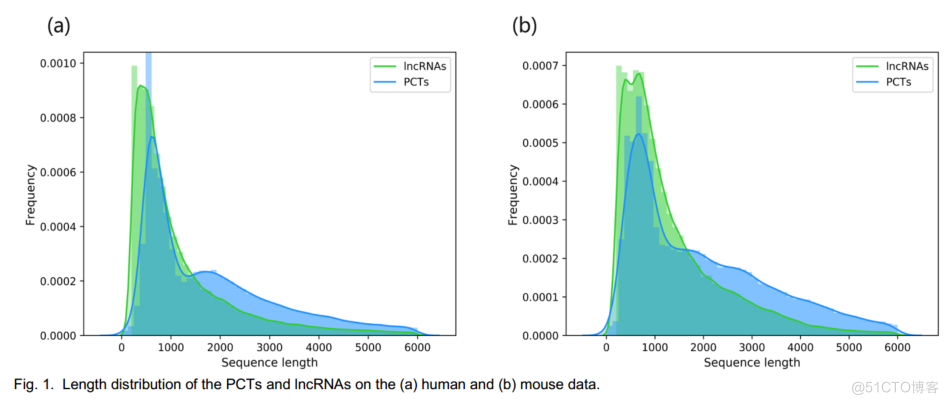
在我们的实验中,只使用长度小于3000个核苷酸的转录本来训练模型。
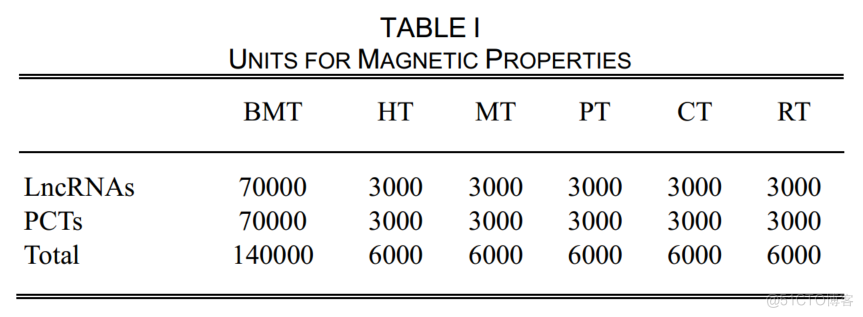
在我们的实验中,我们使用了70000个LncRNA和70000个人类的PCTs来训练人类的模型,这也是训练其他物种识别模型的基本模型。从数据库中删除训练数据集后,分别从NONCODE和GENCODE中随机选择3000个人类LncRNAs和3000个PCTs来评估模型(HT)。为了多样性,我们从NONCODE中制备了小鼠、猪、牛和大鼠的LncRNA。
在表1中,小鼠、猪、牛和大鼠的测试数据集分别记录为MT、PT、CT和RT。
UNITSFORMAGNETICPROPERTIESLNCRNASTABLEI700003000BMT70000300030003000PCTS300030006000300030006000400006000300030006000TOTAL6000MTPTTRTCT
特征提取方法
K-mer频率特征
k-mer方法是由核苷酸(A, G, C, T)组成的长度为k的简单序列。
对于k值较大的k-mer频率特征的分析,通常需要更深层的神经网络来捕获更多的细节。
因此,为了适应不同维度数据的神经网络深度差异,设计了基于hinn的分析仪。
在我们的研究中,5-mer频率特征对应的神经网络最深,4-mer频率特征次之,3-mer频率特征最少。后来的实验也验证了该设计的有效性。
快速傅里叶变换和基于3的周期性
快速傅里叶变换是离散傅里叶变换的快速实现。它改进了基于奇、偶、虚、实性质的DFT算法。在我们的研究中,FFT的引入解决了二进制序列过于稀疏的问题。
基于3的周期性,即包含蛋白质编码区的长度为N的DNA/RNA序列的傅立叶功率谱曲线的频率N/3处出现明显的峰值。已证明DNA/RNA序列中的基于3的周期性主要是由编码区核苷酸分布不平衡引起的。外显子区三个密码子位置的核苷酸分布不平衡,而内含子区域三个密码子位置的核苷酸分布均匀。
在我们的工作中,将原始频谱特征直接输入到模型中。基于CNN的检测器被用来挖掘更有价值的信息。
1)基于cnn的检测器
给定长度为L的转录组序列。第一步是用(1)对序列进行二值化。
N=0,L,2,..,L-1L,S[N]=B0S[N大BLB-ORI
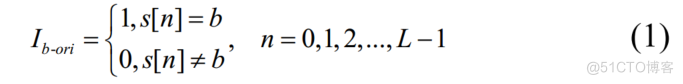
其中
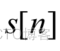
表示转录组序列,

为离散二值时间序列,
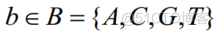
。
为了便于神经网络模型的训练,将四个离散二值时间序列LHOM,LCOANSLOOM,LTAOI
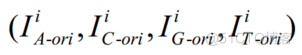
用“0”填充至相同的长度3000。然后,我们对每个离散二值时间序列进行FFT,得到其在频域的表示。
本文给出了离散傅里叶变换的公式。
F[K=>L.[LEK=0.1.2…..2999.2元J230002999水一
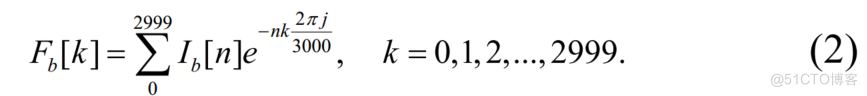
由于光谱是对称的,所以只使用了光谱的前半部分。此外,不直接将光谱作为特征,而是将实部和虚部作为两个通道。从而获得形状为(4,1500,2)的转录组序列的频谱特征。
如前所述,cnn是处理频谱特征方便高效的神经网络。针对转录组序列数据光谱特征的独特特点,我们设计了一种基于cnn的检测器。
由于频谱特征有两个通道,我们首先使用二维卷积提取局部特征。
“A”、“C”、“G”和“T”核苷酸在序列中同样重要。因此,在每个卷积步骤中,同时提取所有核糖核苷酸的局部频谱特征。
因此,我们设计了(4,4),(4,8)和(4,16)三种尺寸的过滤器,每种类型有16个。在Keras中Conv2D图层的超参数“padding”被设置为“valid”。经过二维卷积处理和二维最大池化层降维后,三维特征图生成为二维特征图。接下来,使用一维卷积层和一维最大池化层进一步进行特征提取。最后,经过几个隐藏层处理,我们得到了一个可以表示转录组序列变化的8维向量
2)基于hinn的分析仪
实验发现,3-mer、4-mer和5-mer频率特征比其他模式的识别效果更好。本文设k = 3,4,5。有=134444+454
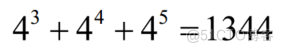
模式,步长设置为1个核苷酸。对于长度为L的序列,由式(3)可计算出1344个k-mer频率特征。
I=1.2,…,1344S=L-K+L,F=立K=3,4.54)

其中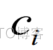 是序列中k-mer频率特征的个数,
是序列中k-mer频率特征的个数, 是其中一个特征。基于hinn的分析仪专门用于分析和提取k-mer频率特征的有效信息。
是其中一个特征。基于hinn的分析仪专门用于分析和提取k-mer频率特征的有效信息。
MERGELAYERCONNECTEDLAYEFREQUENCYFREQUENCYTREQUENCY5-MCR3-MCR4-MCRFULLYOFF非市老移市老专市卡000-+44参电电电善中号电电8电电楼66电电666电电电电中中中

3) 损失函数
选择的损失函数为在标准交叉熵准则中添加一个调制因子
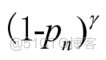
的焦点损失函数,参数为正数,调制因子迫使模型聚焦于难、误分类的样本,焦点损失表示为:
()=–2((IP)R(1P,)’YLOG(P.,)+P'(1-Y,)LOG(-P,))BATCHN=(5
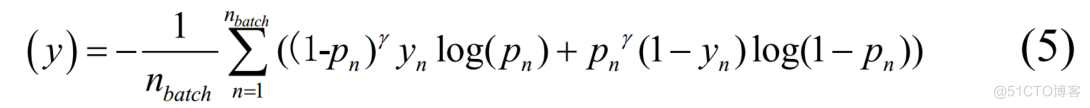
其中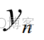 是真实标签,
是真实标签,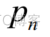 是预测概率,
是预测概率,

是批次大小。
3主要实验及结果
A.性能预测
通过各种超参数组合和大量对比实验,建立了经验最优超参数和模型结构。在本节中,所有的实验结果都是基于BMT的五折交叉验证得到的平均性能。
表2显示了每个组件的模型性能。
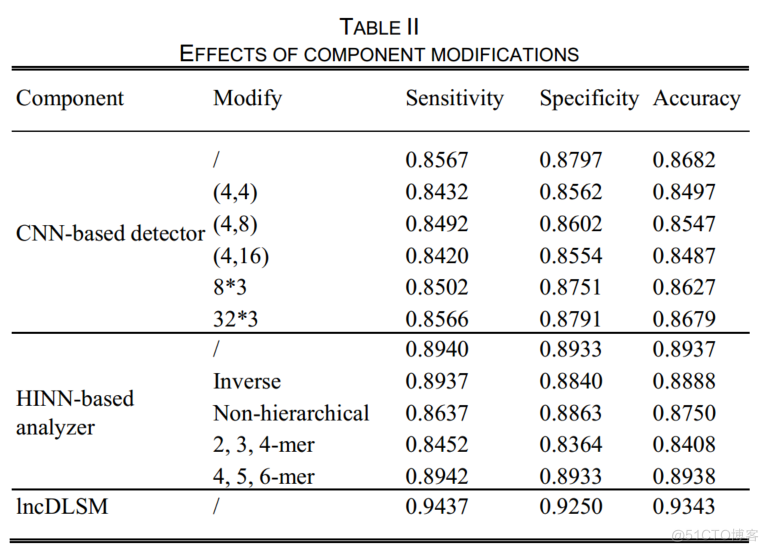
从实验结果可以看出,当使用同一尺寸的过滤器时,过滤器尺寸太大或太小都会变得更糟。这种观察可以从两种极端情况得到直观的解释:一种情况下,过滤器的大小为(1,1),模型不能探索不同特征之间的交叉信息;另一种情况下,过滤器大小等于输入分辨率,卷积层变成全连通的层,这在精度和效率上存在劣势。如上所述,具有多尺度滤光片的卷积层因其优异的实验性能而逐渐被单尺度滤光片所取代。与单尺度滤波器相比,由于使用了多尺度滤波器,基于CNN的检测器同样获得了更令人满意的结果。结果表明,卷积滤波个数为163时精度最高。
对于基于Hinn的分析器(使用3-mer、4-mer、5-mer;5-mer对应于最深的神经网络),
我们进行了以下修改:
1颠倒k-mer的顺序(3-mer对应于最深的神经网络);
2去除层次(hierarchical)结构;
3分别用2、3、4-mer和4、5、6-mer取代3、4、5-mer。
4与基于非层次的Hinn分析器相比,未修改和反向(inversed )的基于Hinn的分析器都获得了更高的准确率,证实了分层输入的必要性。实验结果表明,基于Hinn的分析器设计的合理性是基于人类认知过服务器托管网程的。
5在特征替换实验中,4、5和6-mer频率特征获得了最好的结果,略有优势。在准确率和效率的权衡下,我们选择了3、4和5个频率特征来达到识别的目的。
6LncDLSM包括基于CNN的检测器和基于Hinn的分析仪,取得了最好的识别效果。
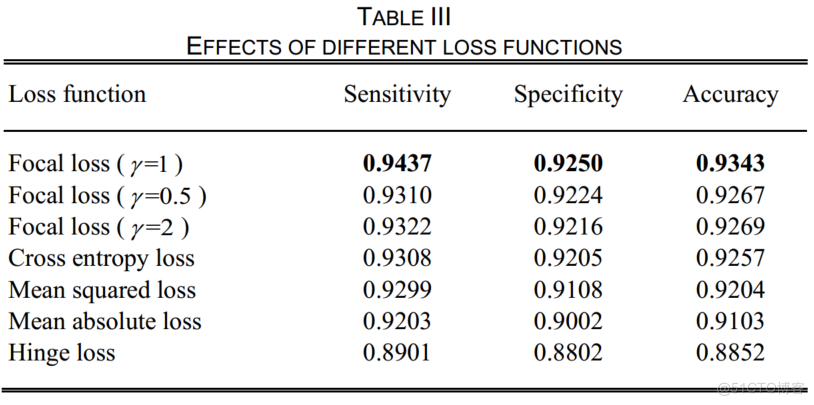
表3显示了一个明显的结论,即焦点损失是该模型的最佳目标函数。焦点损失集中在难的、错误分类的样本上,以促进模型参数更接近全局解。在实验中,值为1时精度最高。
B.预测方法的比较
为了验证模型的有效性,将LncDLSM与以下识别方法进行了比较:
1基于神经网络的LncRNAnet模型使用RNN学习序列的内在特征,并提出了一个ORF指示器来加强模型;
2CPAT提取ORF大小、ORF覆盖率、Fickett TESTCODE统计和六角体使用偏差四个序列特征来建立Logistic回归模型;
3CPC2是使用了由六个具有生物学意义的序列特征构建的支持向量机(SVM)模型,包括对数赔率分数、ORF覆盖、ORF完整性、命中次数、命中分数和帧分数;
4PLEK,一个基于改进的k-mer方案的无对齐工具,也是一个基于支持向量机的模型,具有径向基函数;
5mRNN使用GRU来解决由长核酸序列引起的“消失梯度问题”。
6RNAsamba,一种基于神经网络的模型。从初始核苷酸序列开始,RNAsamba考虑了来自两个不同来源的信息,整个核苷酸序列和最长的ORF,以计算给定转录本的编码潜力。
1)人类数据集的性能比较
表4显示了在人类测试数据集上的识别性能。
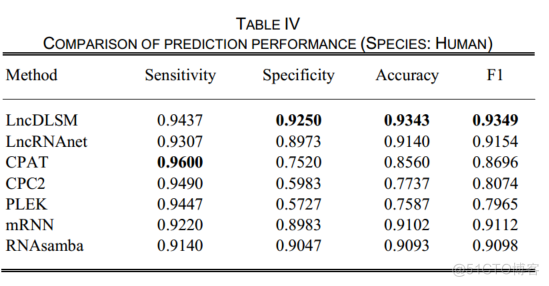
1LncDLSM的预测准确率为93.43%,分别比lncRNAnet、CPAT、CPC2、PLEK、mRNN和RNAsamba的预测准确率高2.03%、7.83%、16.06%、17.56%、2.41%和2.50%。并且lncDLSM在特异性和F1方面的得分最高。
2虽然CPAT具有最高的敏感性,但它的特异性、准确性和F1较差。CPC2和PLEK在各方面的表现相似。此外,CPAT、CPC2和PLEK的预测存在偏向于lncRNAs的现象。
3LncRNAnet对PCTs的预测相对较好,但并没有完全解决问题。一种可能的解释是,许多lncRNAs具有与PCTs相似的ORF。LncRNAnet通过CNN检测ORF指示符,而不是计算ORF的统计特性,在一定程度上提高了伪ORF的识别能力。此外,lncRNAnet通过RNN学习固有特征,进一步增强了模型的识别能力。PLEK忽略了序列的变异,导致特异性最低。相比之下,LncDLSM获得了最好的总体性能,并平衡了敏感性和特异性。此外,最高的F1也证明了这一点。
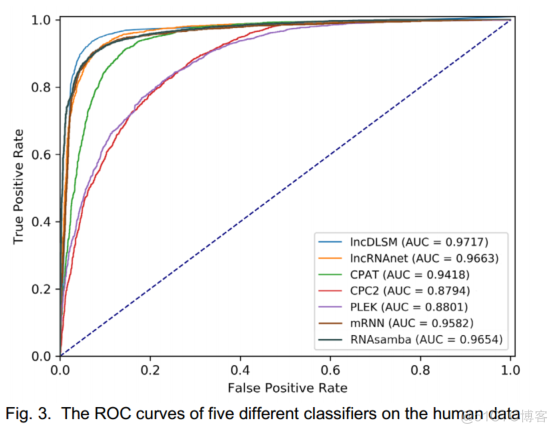
在图3中,我们绘制了ROC曲线,并计算了AUC。lncDLSM(0.9717)获得了最高的AUC得分。这进一步证实了上述发现,并表明lncDLSM是一个很好的分类器。
2)小鼠数据集性能比较
表5表明,LncDLSM在小鼠数据上仍然取得了良好的性能,具有平衡的灵敏度和特异度。
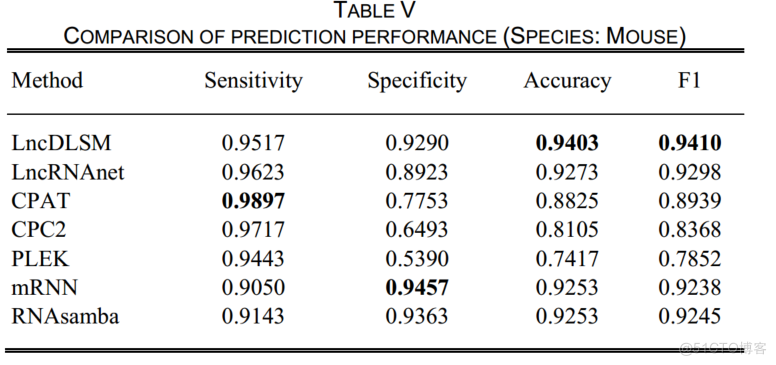
特别是,lncDLSM获得了最高的特异性、准确性和F1,并在敏感性和特异性之间取得了平衡的综合表现。
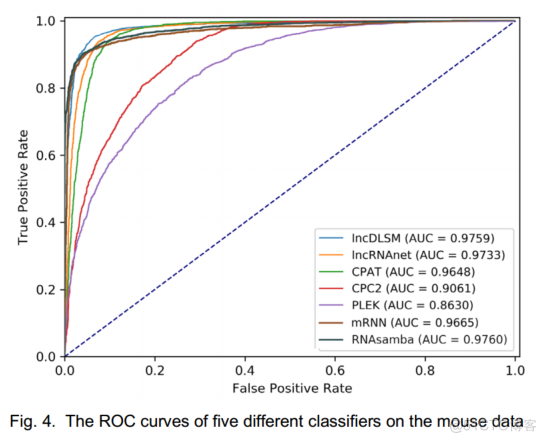
如图4所示,0.9759的AUC分数证明了lncDLSM是一个很好的分类器.
3)其他数据集的性能比较
表6列出了与其他物种的实验结果。
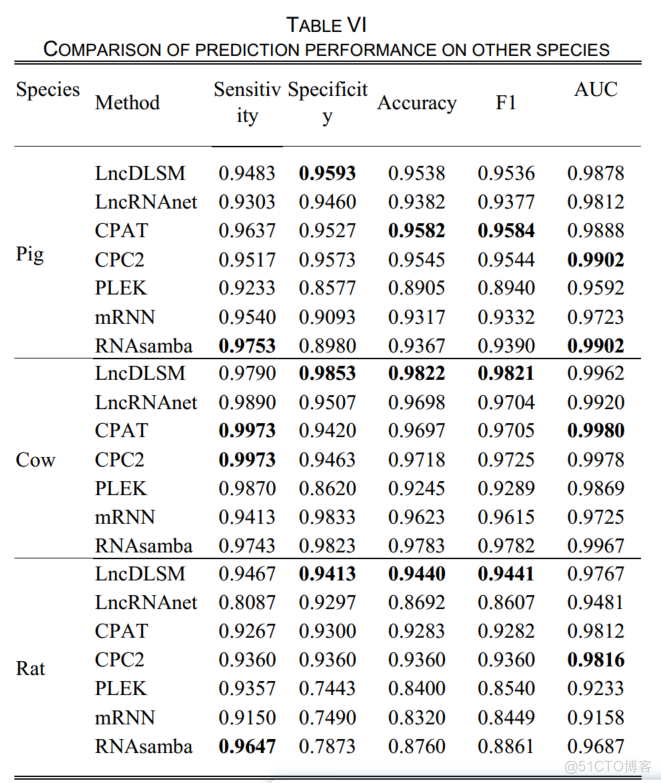
LncDLSM在敏感度、特异度、准确性、F1和AUC方面表现良好。
特别是,LncDLSM在猪、牛和老鼠的数据上获得了94.4%以上的准确率、94.4%的F1和0.976的AUC值。
CPAT和CPC2在猪、牛和老鼠的数据上表现出平衡的性能。相比之下,CPAT和CPC2在人类和小鼠数据中表现不佳。有一个可能的原因是,GENCODE中的PCTs比RefSeq中的PCTs有更长的5’和3’非翻译区(UTRs),这导致了对分类器的误判。
结果表明,lncDLSM和lncRNAnet可以忽略UTRs长度的影响,尤其是lncDLSM。
此外,表5和表6反映了迁移学习可以增强lncDLSM的泛化能力。
实验结果表明,从源域(人类数据)到目标域(其他物种数据)的迁移学习是成功的,没有负迁移。可以想象,物种间的基因同源性保证了成功的迁移学习。事实上,物种专一性是各种模型参数细微差别的来源。
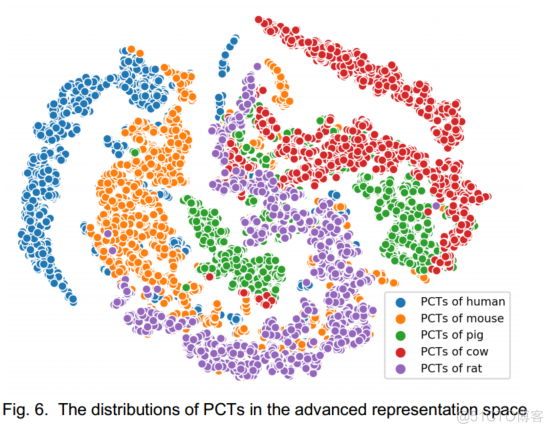
为了进一步解释,我们使用t-SNE对神经网络的最后一层隐藏层的输出进行了降维。
我们根据5个物种的数据绘制了lncRNAs/PCTs的分布。
图5和图6,很容易看出,分布之间的小距离和明显的边界分别对应于物种之间的同源性和特异性。
众所周知,人和小鼠具有很高的DNA同源性。
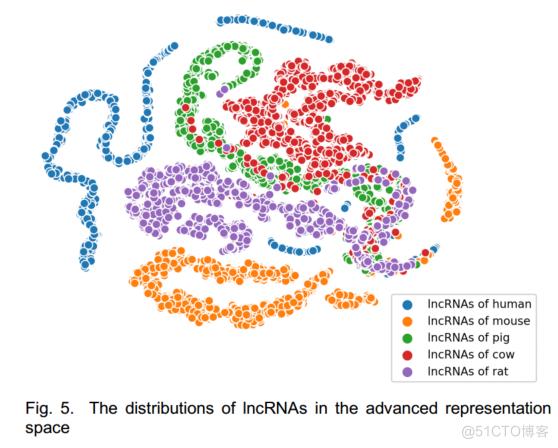
如图6所示,人类和小鼠的PCTs分布之间的距离很小,甚至有轻微的重叠,这是进一步的证据。
若有收获,就点个赞吧
讨论过程
总结
优点
单独的CNN,老师希望能够创新,自己去摸索新方法。
服务器托管,北京服务器托管,服务器租用 http://www.fwqtg.net
机房租用,北京机房租用,IDC机房托管, http://www.fwqtg.net
前言 个人主页: :✨✨✨初阶牛✨✨✨ 推荐专栏: c语言进阶 个人信条: 知行合一 本篇简介:>:介绍c语言中有关指针更深层的知识. 金句分享: ✨今天所有的混乱与芜杂,努力与精进,✨ ✨都将在进步中变得更加清晰.✨ @TOC 一、字符指针 我们可以定…
