
1. 说明 (来自官方文档):
The TissueEnrich package is used to calculate enrichment of tissue-specific genes in a set of input genes.
Tissue-specific genes were defined by processing RNA-Seq data from the Human Protein Atlas (HPA) (Uhlén et al. 2015), GTEx (Ardlie et al. 2015), and mouse ENCODE (Shen et al. 2012) using the algorithm from the HPA (Uhlén et al. 2015).
The hypergeometric test is being used to determine if the tissue-specific genes are enriched among the input genes. Along with tissue-specific gene enrichment, the TissueEnrich package can also be used to define tissue-specific genes from expression datasets provided by the user, which can then be used to calculate tissue-specific gene enrichments.
TissueEnrich has the following three functions:
teEnrichment: Given a gene list as input, this function calculates the tissue-specific gene enrichment using tissue-specific genes from either human or mouse RNA-Seq datasets.
teGeneRetrieval: Given gene expression data across tissues, this function defines tissue-specific genes by using the algorithm from the HPA.
teEnrichmentCustom: Given a gene list and tissue-specific genes fromteGeneRetrievalas input, this function calculates the tissue-specific gene enrichment.
详情见:https://bioconductor.org/packages/release/bioc/vignettes/TissueEnrich/inst/doc/TissueEnrich.html
2. 参考:
TissueEnrich包: https://bioconductor.org/packages/release/bioc/html/TissueEnrich.html
参考文献: Jain A, Tuteja G. TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics. 2019 Jun 1;35(11):1966-1967.
安装教程: https://github.com/Tuteja-Lab/TissueEnrich
3. 目的:
本文只是想对teEnrichment函数得到的富集结果做进一步处理:将一组目标基因集合作为 TissueEnrich 包的输入,利用teEnrichment方法得到其中的组织特异性基因的富集结果。官方给出了某个组织中富集到的基因在所有组织中的表达情况 (如下图所示),这样一次只能获取一个组织的结果,如果要一次获取全部组织的结果,那么可能需要自己手动处理。所以本文旨在提供一种一次性获取所有组织中的特异性基因富集结果 (这些特异性基因限定于 目标基因集合中) 的方法。
下方脚本主要做了两方面处理:1. 将目标基因集合中那些存在组织特异性基因富集结果的取出;2. 对于那些不存在富集结果的目标基因,用 0.0 进行填充 (表示无结果)。最终得到所有目标基因在所有组织中的特异性表达富集结果。
数据来源:
作者并没有将不同来源的数据进行整合,所以用户可以通过rnaSeqDataset指定不同来源的数据 (默认 RNA-Seq 数据用的是HPA的,如下图所示)。


比如:人类Placenta (胎盘)中特异性基因在35个组织中的表达情况 (官方示例)。
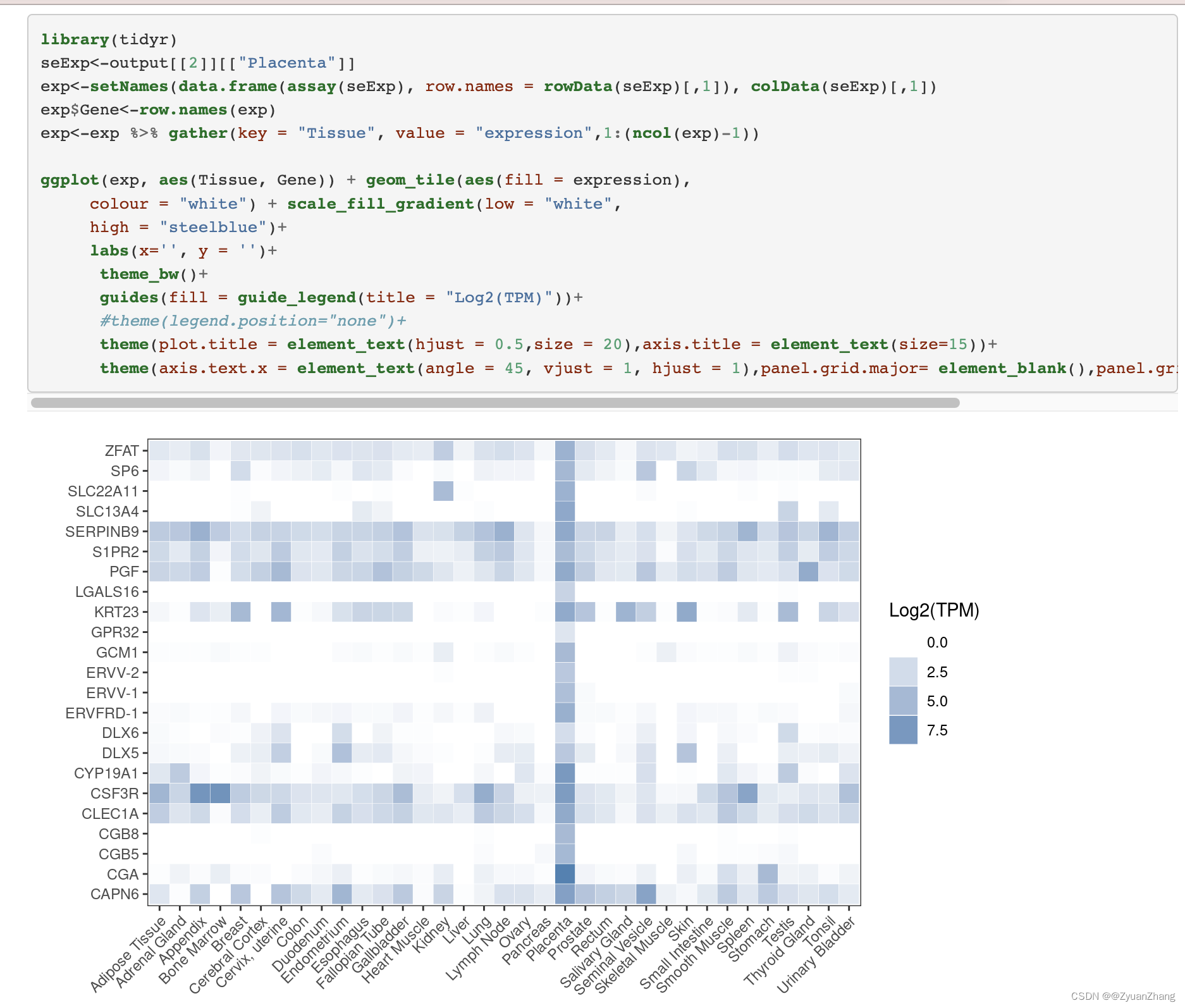
4. 脚本:
主要思路就是先根据 teEnrichment 方法获取目标基因的组织特异性富集结果,之后for循环遍历富集结果中的每一个组织的特异性基因富集结果。富集结果中,某些目标基因可能不存在,那么就用0.0填充(即 该基因在所有组织中不表达)。最终将所有结果拼接输出,得到所有目标基因的组织特异性富集结果。
library(TissueEnrich)
library(tidyr)
library(dplyr)
getTargetGeneExpression = function(inputGenes, organism, outfilePath, rnaSeqSource=1, ifHeatMap=FALSE){
# 获取所有目标基因在人体组织中的表达分布
## 1. 获取特定组织中的基因富集结果
getExpress = function(tissuename) {
expressdata = output[[2]][[tissuename]]
return(expressdata)
}
## 2. 绘制热图的函数
drawHeatMap = function(expdf) {
ggplot(expdf, aes(Tissue, Gene)) +
geom_tile(aes(fill = expression),
colour = "white") +
scale_fill_gradient(low = "white",
high = "steelblue")+
labs(x='', y = '')+
theme_bw()+
guides(fill = guide_legend(title = "Log2(TPM)"))+
#theme(legend.position="none")+
theme(plot.title = element_text(hjust = 0.5,size = 20),
axis.title = element_text(size=15))+
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank())
}
## 根据基因列表用 teEnrichment() 进行富集
gsGeneSet(geneIds=inputGenes, organism=organism, geneIdType=SymbolIdentifier())
outputteEnrichment(inputGenes=gs, rnaSeqDataset=rnaSeqSource)
enrich_gene_dataframe = data.frame(matrix(nrow=0, ncol=length(rownames(output[[1]]))+1)) ## 有富集结果的基因
not_enrich_gene_dataframe = data.frame(matrix(nrow=0, ncol=length(rownames(output[[1]]))+1)) ## 没有富集结果的基因
colnames(enrich_gene_dataframe) = c(rownames(output[[1]]), "Gene")
colnames(not_enrich_gene_dataframe) = c(rownames(output[[1]]), "Gene")
## 提取有富集结果的基因的表达
for (tissuename in rownames(output[[1]])) {
result = getExpress(tissuename)
if (dim(result)[1] != 0) {
exp = setNames(data.frame(assay(result), row.names = rowData(result)[,1]), colData(result)[,1])
exp$Gene = row.names(exp)
enrich_gene_dataframe = rbind(enrich_gene_dataframe, exp)
}
}
enrich_gene_dataframe = distinct(enrich_gene_dataframe, .keep_all = TRUE) ## 去掉完全重复的行
## 提取没有富集结果的基因,并用 0 填充
not_enrich_gene = setdiff(inputGenes, enrich_gene_dataframe$Gene)
for (ng in not_enrich_gene) {
result = rep(0.0, ncol(not_enrich_gene_dataframe)-1)
result = data.frame(t(as.data.frame(result)))
result$Gene = ng
colnames(result) = colnames(not_enrich_gene_dataframe)
not_enrich_gene_dataframe = rbind(not_enrich_gene_dataframe, result)
}
result_dataframe = rbind(enrich_gene_dataframe, not_enrich_gene_dataframe)
order_result_dataframe = result_dataframe[order(result_dataframe$Gene),] ## 根据 Gene 名称排序
write.table(order_result_dataframe, file=outfilePath, quote=FALSE, row.names=FALSE, sep="t")
if (ifHeatMap) {
order_result_dataframe = order_result_dataframe %>% gather(key = "Tissue", value = "expression",1:(ncol(order_result_dataframe)-1))
drawHeatMap(order_result_dataframe)
}
}
5. 运行方式
#------输入数据------#
gene_dataframe = read.csv("your_target_gene_files.txt", sep="t", header = TRUE)
inputGenes = gene_dataframe$Gene ## 获取 Gene 所在的列 (此处输入文件中 Gene 所在列的名称为 "Gene")
organism = "Homo Sapiens" ## 物种 (此处选的是 人)
rnaSeqSource = 2
# An integer describing the dataset to be used for enrichment analysis.
# 1 for 'Human Protein Atlas' (default),
# 2 for 'GTEx',
# 3 for 'Mouse ENCODE'. Default 1.
outfilePath = "your_result.txt" ## 所有目标基因的组织富集结果 (输出文件)
getTargetGeneExpression(inputGenes, organism, outfilePath, rnaSeqSource, ifHeatMap=TRUE)
## 关于 getTargetGeneExpression() 函数中各参数的含义:
## inputGenes : 输入的基因集合,可以读取文件中的基因集合所在的列;
## organism: 物种 (只有 人(Homo Sapiens)和鼠(Mouse) 两种选择)
## outfilePath: 输出文件
## ifHeatMap: 是否绘制热图 (默认不绘制,即 ifHeatMap=FALSE)
6. 示例:
以官方给出的基因集合为例:
genessystem.file("extdata", "inputGenes.txt", package = "TissueEnrich")
inputGenesscan(genes,character())
rnaSeqSource = 1 ## 可以不写,因为默认是 1
organism = "Homo Sapiens"
outfilePath = "example_result.txt"
getTargetGeneExpression(inputGenes, organism, outfilePath, ranSeqSource, ifHeatMap=TRUE)
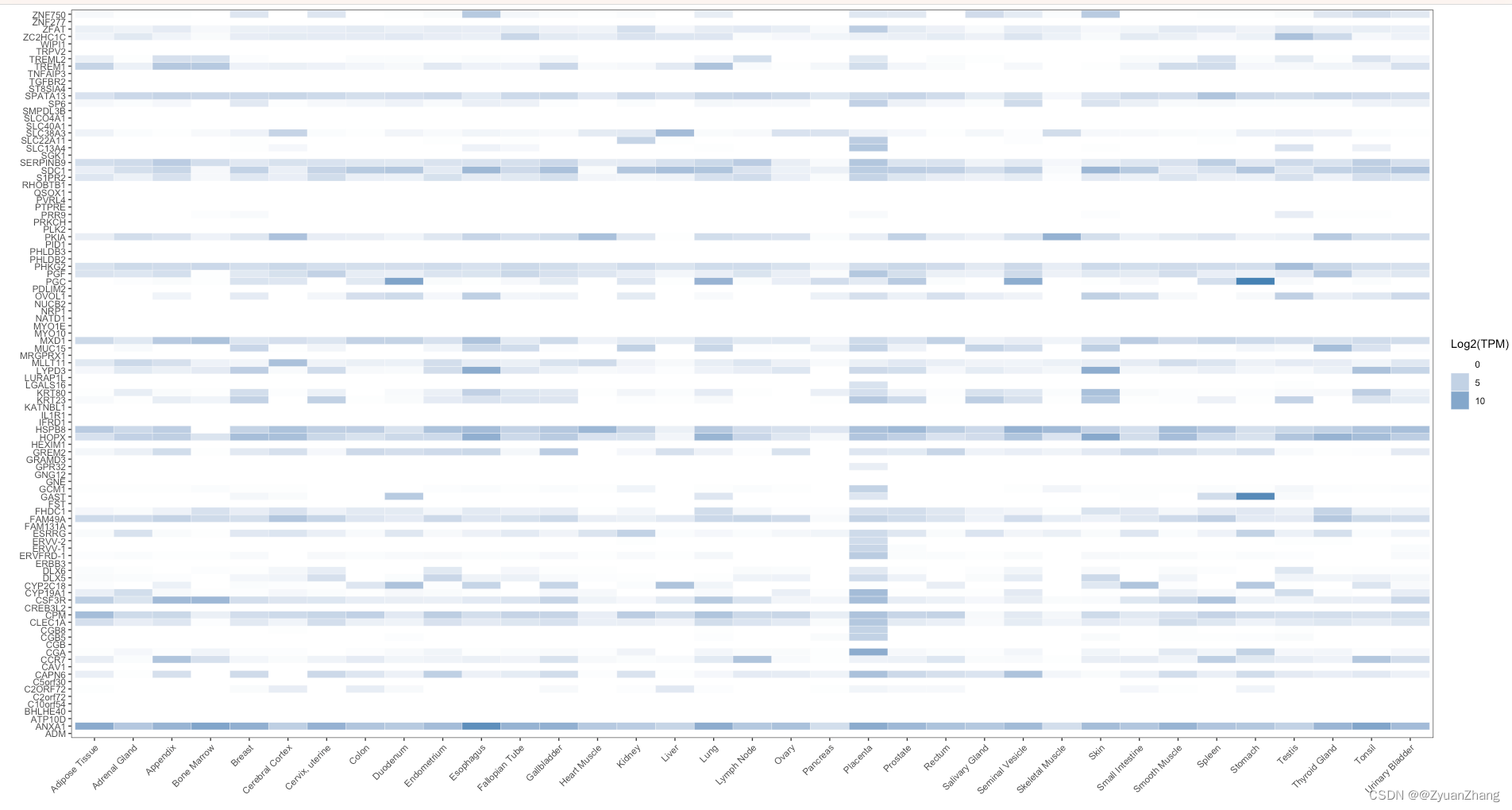
结果如下:
一共有97个基因,其部分富集结果如下:
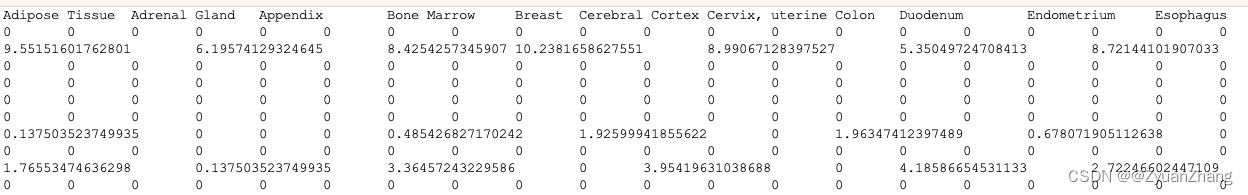
可视化热图结果如下:
服务器托管,北京服务器托管,服务器租用 http://www.fwqtg.net
